

En octobre 2022, le GP2 a annoncé la troisième édition de la publication de données sur la plateforme Terra, en collaboration avec AMP® PD. Cette publication regorge de données et de ressources inédites et comprend presque deux fois plus d’échantillons que la publication précédente.
Cette édition comprend 6 330 participants de plus dans la catégorie des maladies complexes, qui viennent s’ajouter aux éditions précédentes issues des réseaux de maladies complexes et monogéniques. Les données sur les maladies complexes totalisent désormais 14 902 participants au génotype référencé (8190 MP et 6,712 non-MP). Les nouvelles cohortes de cette édition sont les suivantes :
- Étude australienne sur la génétique de Parkinson (APGS)
- Fox Investigation for New Discovery of Biomarkers (BioFIND), une étude d’observation clinique de la maladie de Parkinson
- Sites participants provenant de la cohorte MP de sujets noirs et afro-américains, Black and African American Connections to Parkinson’s Disease (BLAACPD-KPM, BLAACPD-RUSH, BLAACPD-UAB, BLAACPD-UC)
- Cohorte du GP2 de l’Université Koc (KOC), une cohorte basée en Turquie
- Consortium de cohorte LRRK2 (LCC), une cohorte de MP de porteurs du gène de mutation LRRK2
- Génotypes et phénotypes des troubles moteurs – King’s College London (MDGAP-KINGS), une banque de cerveau du Royaume-Uni
- Programme Parkinson de Nouvelle-Zélande (NZP3)
- Étude sur la génétique de Parkinson et l’environnement (PAGE)
- Initiative sur les marqueurs de progression de Parkinson (PPMI)
- Étude systémique des échantillons de synucléine (S4), une étude des biomarqueurs de la MP sur l’α-synucléine.
Le lignage génétiquement déterminé de la maladie complexe chez les participants du GP2 se divise en dix lignées (les neuf lignées ci-dessous et un petit nombre de Finlandais d’Europe). Le tableau ci-après présente la lignée génétiquement déterminée des participants à cette édition, atteints de maladies complexes, ayant passé le contrôle de qualité et pris en compte. Ces chiffres comprennent les échantillons des éditions précédentes, reclassés selon une nouvelle nomenclature et soumis à un contrôle de qualité, ainsi que de nouveaux échantillons génotypés et exclusifs à cette édition. 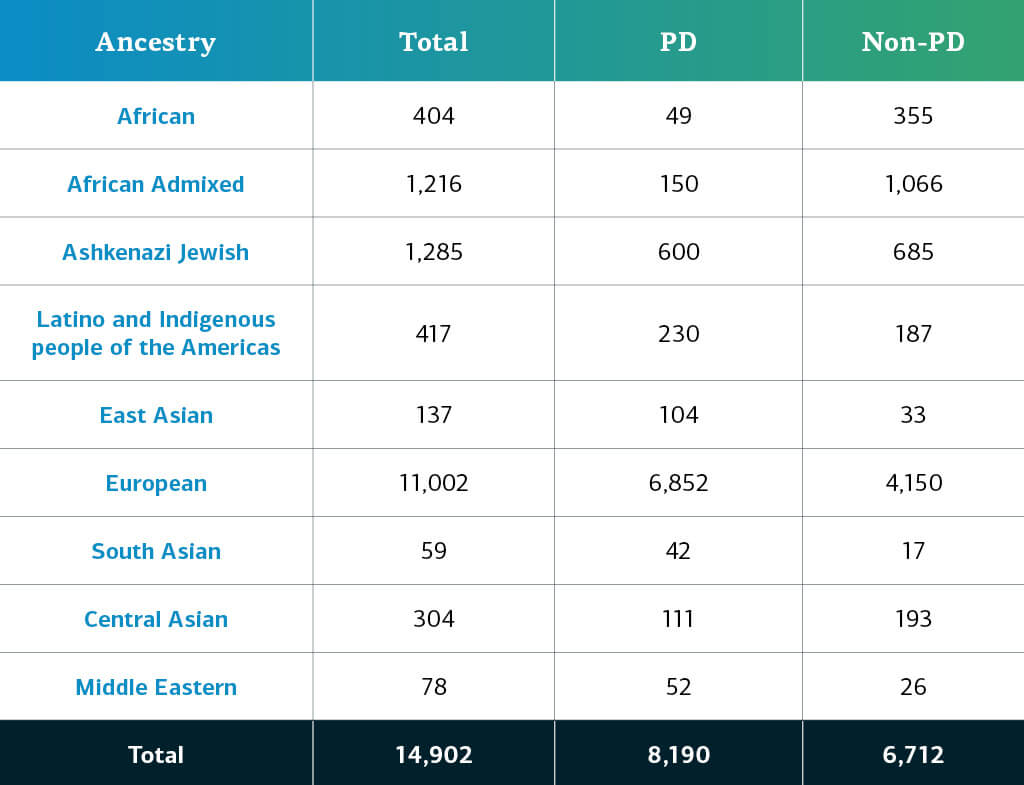 Les futures publications de données contribueront à renforcer la diversité des participants disponibles. Vous pouvez consulter notre tableau de bord pour suivre nos progrès.
Les futures publications de données contribueront à renforcer la diversité des participants disponibles. Vous pouvez consulter notre tableau de bord pour suivre nos progrès.
Le résultat premier de cet ensemble de données est qu’il comprend davantage d’échantillons avec des données génotypés à plus large spectre ainsi que des métadonnées cliniques et génomiques supplémentaires. Ces données ont été regroupées par génotype dans un fichier de classement propre au GP2 (disponible dans les paramètres des répertoires répondant à un accès aux données de niveau 1 et 2). Ce fichier spécifique compte 2793 échantillons et 6 groupes de lignage représentatifs de la diversité génétique des lignées comprises dans cette édition de données et est enrichi de 420 cas de maladie de Gaucher afin de tenir compte des variants génétiques présentant un intérêt en terme de gène GBA à risque. Ce fichier peut également être téléchargé sur le répertoire GitHub du GP2 pour l’application des données hors du cadre du GP2.
Nous avons également mis à jour les entrées du nombre de copies variables (NCV) pour tous les échantillons génotypés soumis au contrôle de qualité (niveau de gène plus 250kb de séquençage des régions flanquantes). Le NCV indique les variations du nombre de copies d’un brin d’ADN donné. Cette variation peut tenir à des délétions, duplications ou autres événements et peut fournir des informations complémentaires sur la façon dont la variation structurelle affecte le risque de maladie. Les outils utilisés pour produire des séquences probables de NCV sont disponibles sur le GitHub du GP2. Ces résultats sont regroupés par lignage génétique dans l’espace de travail de niveau 2. Ces NCV de probabilité sont un excellent point de départ pour prioriser les échantillons porteurs d’insertions, de délétions ou de duplications potentielles dans des gènes représentant un intérêt pour les études de suivi et autres analyses.
Des informations complémentaires sur la structure du génotype des maladies complexes et sur les données cliniques sont disponibles dans l’article «Les éléments qui composent la première édition des données du GP2»’ ainsi que dans le document README, mis à jour pour cette édition et disponible dans les espaces de travail officiels du GP2 sur Terra. Les données monogéniques sur la maladie de Parkinson-séquençage de génome complet (PD WGS) sont également disponibles dans ce même document README. D’autres données sur le séquençage de génome complet monogénique seront publiées à l’occasion de la prochaine édition.
L’aperçu de la prochaine édition comprend (visant la période de congés, calendrier 1er trimestre 2023) :
- Autres données de génotype
- Autres données sur le séquençage de génome complet (WGS)
- Autres métadonnées cliniques
- Code et outils d’analyse mis à jour
- Autres résumé des statistiques portant sur le risque et l’âge au démarrage des études d’association pangénomiques
Les auteurs de ce blog sont membres duGroupe de travail et d’analyse de données – maladies complexes: Hampton Leonard, Mike Nalls, Dan Vitale et Mary Makarious ; Matthew Korestsky du National Institutes of Health ; Kristin Levine de Data Tecnica International/National Institutes of Health ; Zih-Hua Fang et Peter Heutink, membres du Groupe de travail sur l’analyse des données monogéniques.



