Im April 2022 kündigte GP2 in Zusammenarbeit mit AMP ® die zweite Datenveröffentlichung auf der Terra-Plattform an PD. Dieser Release enthält Daten aus den beiden GP2-Netzwerken Komplexe Krankheit und Monogene Krankheit. Die Daten zur komplexen Krankheit umfassen nun insgesamt 8644 genotypisierte Teilnehmer*innen (5249 mit Parkinson-Erkrankung, 3395 ohne). Neu hinzugekommen bei dieser Freigabe sind die Movement Disorders Genotypes and Phenotypes – Queen Square Brain Bank (MDGAP-QSBB), eine Hirnbank aus dem Vereinigten Königreich, und SYNAPS Study – Kazakhstan (SYNAPS-KZ), eine Parkinson-Kohorte aus Kazakhstan. Auch weitere CORIELL-Proben sind mit dieser Freigabe hinzugekommen. Die Daten zu monogenen Erkrankungen umfassen 235 Teilnehmer mit PD aus der PDGENEration- Kohorte, bei denen das gesamte Genom sequenziert wurde (WGS). , die nach festgelegten Kriterien ausgewählt wurden für den Verdacht auf eine monogene Ursache der Parkinson-Krankheit vom Netzwerk für monogene Erkrankungen.
Der Hauptunterschied zwischen Genotypisierungs-Microarray- und WGS-Daten liegt in der Anzahl der genetischen Marker, die während des Genotypisierungsprozesses erkannt werden. WGS (Whole Genome Sequencing oder Gesamtgenomsequenzierung) liefert einen umfassenden Überblick über das Genom, indem potenziell alle 3,2 Milliarden Basenpaare im Humangenom abgefragt werden, während bei der Genotypisierung mit einem gezielten Microarray (wie dem speziellen NeuroBooster-Array von GP2) eine gezieltere Anzahl von bis zu 1,9 Millionen Region-Tagging-Markern pro Probe abgefragt wird. WGS eignen sich besser für Analysen zur Untersuchung seltener genetischer Variationen, während Genotypen, die einem gut passenden Referenzpanel zugeschrieben werden, eine skalierbare und effiziente Lösung für die Untersuchung häufiger genetischer Variationen darstellen.
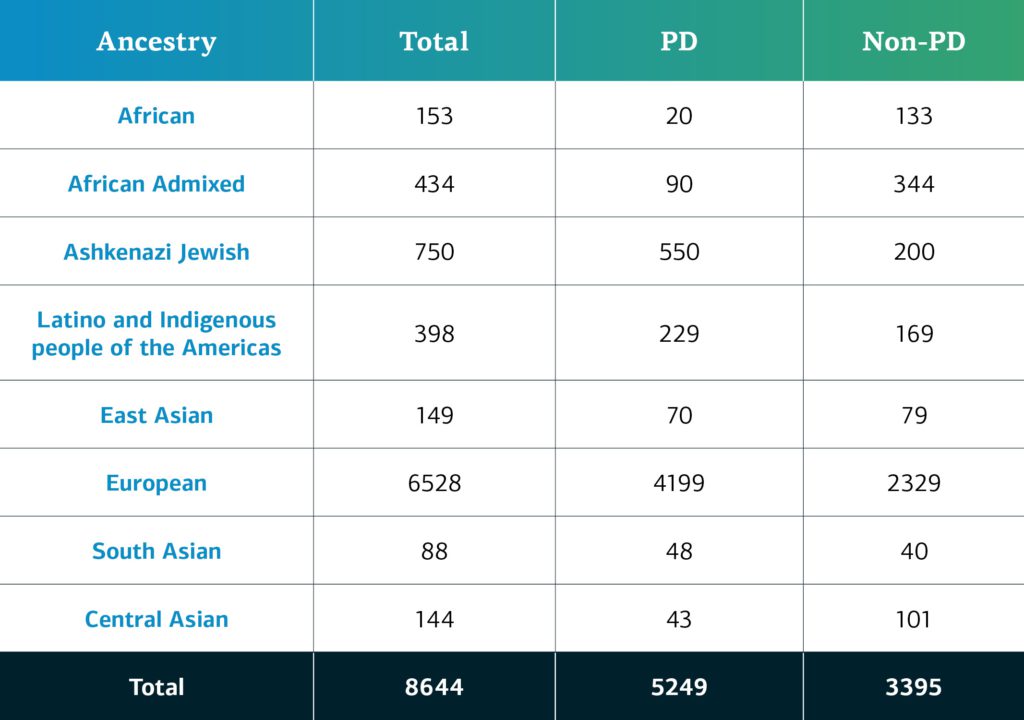
Die genetisch bedingte Abstammung von Teilnehmern an GP2 mit komplexen Erkrankungen ist in neun Abstammungsgruppen unterteilt. In der nachstehenden Tabelle sind die genetisch bedingten Abstammungen von Teilnehmern an komplexen Erkrankungen in GP2 Release 2 aufgeführt, die die Qualitätskontrolle bestanden und imputiert wurden.
Die genetisch bestimmte Abstammung der Teilnehmer an der monogenen Krankheit GP2 wird derzeit in vier Hauptabstammungsgruppen unterteilt, da dies die ersten verfügbaren Daten aus dem monogenen Netzwerk sind. Im Zuge der Verarbeitung weiterer Daten werden vielfältigere Proben zur Verfügung stehen. In der nachfolgenden Tabelle sind die genetisch bedingten Vorfahren der Teilnehmer an monogener Krankheit in GP2 Release 2 nach Erkrankungsalter und Familienanamnese aufgeführt.
<img class="aligncenter wp-image-50009472" src="https://gp2.org/wp-content/uploads/2022/05/MicrosoftTeams-image-42-1024×426.jpg" alt="Eine Datentabelle, die die Beziehung zwischen Familiengeschichte, Abstammung und Alter bei Ausbruch einer Erkrankung zeigt. Die Spalten sind nach den Kategorien „Alter bei Ausbruch“ beschriftet: „ 60“, mit weiterer Unterteilung in „Ja“ und „Nein“ für die Familiengeschichte. Die aufgeführten Abstammungsgruppen umfassen europäisch, asiatisch und gemischt.“ width=“844″ height=“351″ />
Durch zukünftige Datenfreigaben wird sich die Vielfalt der verfügbaren Teilnehmenden weiter erhöhen. Sie können sich unseren Fortschritt auf unserem Dashboard ansehen .
Zusätzlich zur Aufnahme der ersten WGS-Daten haben wir einen weiteren neuen Datentyp aufgenommen: Die probabilistische Kopienzahlvariante (CNV) erfordert, dass alle genotypisierten Proben die Qualitätskontrolle bestehen (Genebene plus 250 kb flankierende Regionen). Die CNV beschreibt die Variation in der Anzahl der Wiederholungen eines bestimmten DNA-Abschnitts. Diese Variation kann durch Deletion, Insertion oder andere Ereignisse entstanden sein und liefert möglicherweise mehr Informationen darüber, wie strukturelle Variationen das Krankheitsrisiko beeinflussen. Die Pipeline, die zur Erzeugung der probabilistischen CNV-Aufrufe verwendet wird, finden Sie auf dem GP2 Github . Dies ist derzeit in Arbeit und wird verbessert, wenn wir mehr Daten einbeziehen und Anpassungen an der Pipeline vornehmen. Betrachten Sie diese CNV-Daten als „Hypothesengenerierung“. Hinweise zur Verwendung werden im Blogbeitrag zur ersten stabilen Version enthalten sein, die für das nächste Quartal geplant ist.
Diese Version enthält neben den genotypisierten Daten zu komplexen Erkrankungen des NeuroBooster-Arrays auch WGS-Daten aus dem Netzwerk für monogene Erkrankungen. Weitere Informationen zur Struktur des komplexen Krankheitsgenotyps und zu klinischen Daten finden Sie im Blogbeitrag „ Die Komponenten der ersten Datenveröffentlichung von GP2 “. sowie in der README-Datei, die bei jeder Veröffentlichung aktualisiert wird und in den offiziellen GP2 Terra-Arbeitsbereichen verfügbar ist. Die monogenen PD-WGS-Daten werden ebenfalls in derselben README-Datei ausführlich beschrieben.
Wir freuen uns sehr, der Parkinson-Forschungsgemeinde diesen Beta-Release zur Verfügung stellen zu können – und es kommt noch viel mehr!
Dieser Blog wurde gemeinsam von Hampton Leonard, Mike Nalls, Dan Vitale, Yeajin Song, Kristin Levine, Mary Makarious, Zih-Hua Fang und Peter Heutink verfasst. Bitte besuchen Sie die Arbeitsgruppen „Complex Disease – Data Analysis“ und „Monogenic – Data Analysis Working Group“ von GP2, um mehr über deren Hintergrund zu erfahren.
Schauen Sie sich unsere anderen Datenveröffentlichungen an.