En avril 2022, le GP2 a annoncé la diffusion du deuxième ensemble de données sur la plateforme Terra en collaboration avec AMP® PD. Cette diffusion contient des données provenant à la fois des réseaux du GP2 sur les maladies complexes et sur les maladies monogéniques. Les données sur les maladies complexes comptent désormais un total de 8 644 participants génotypés (5 249 MP, 3 395 non-MP). Les nouveautés de cette diffusion sont : génotypes et phénotypes des troubles moteurs – Queen Square Brain Bank (MDGAP-QSBB), une banque de cerveaux du Royaume-Uni, et SYNAPS Study – Kazakhstan (SYNAPS-KZ), une cohorte MP du Kazakhstan. Des échantillons CORIELL supplémentaires ont également été ajoutés à cette édition. Les données sur les maladies monogéniques portent sur 235 participants atteints de la maladie de Parkinson et dont le séquençage de génome complet a été effectué. Ces participants font partie de la cohorte PDGENEration (https://www.parkinson.org/PDGENEration) et ont été sélectionnés conformément aux critères établis sur la suspicion d’une cause monogénique de la maladie de Parkinson.
La principale différence entre le génotypage des données des biopuces et le séquençage du génome complet réside dans le nombre de marqueurs génétiques détectés au cours du processus de génotypage. Le séquençage du génome complet permet une vue exhaustive du génome en interrogeant potentiellement les 3,2 milliards de paires de bases du génome humain, tandis que le génotypage avec une biopuce ciblée (telle que la biopuce NeuroBooster personnalisée du GP2) interroge un nombre plus ciblé de marqueurs moléculaires par région, jusqu’à 1,9 million par échantillon. Les séquençages du génome complet sont mieux adaptés aux analyses portant sur les variations génétiques rares tandis que les génotypes imputés à un panel de référence bien apparié constituent une solution évolutive et efficace pour étudier les variations génétiques plus courantes.
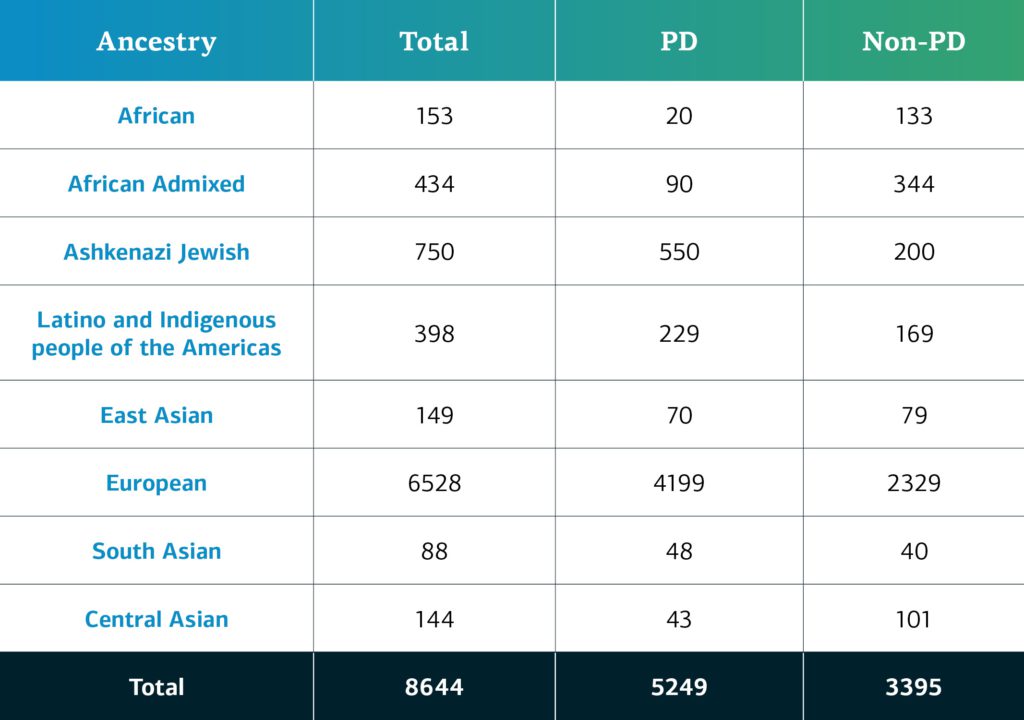
L’ascendance génétiquement déterminée des individus participant à l’enquête GP2 sur les maladies complexes est répartie en neuf groupes d’ascendance. Le tableau ci-dessous précise l’ascendance génétiquement déterminée des individus participant au GP2 dans le cadre de cette deuxième diffusion de données, qui ontt passé le contrôle de qualité et ont été attribués.
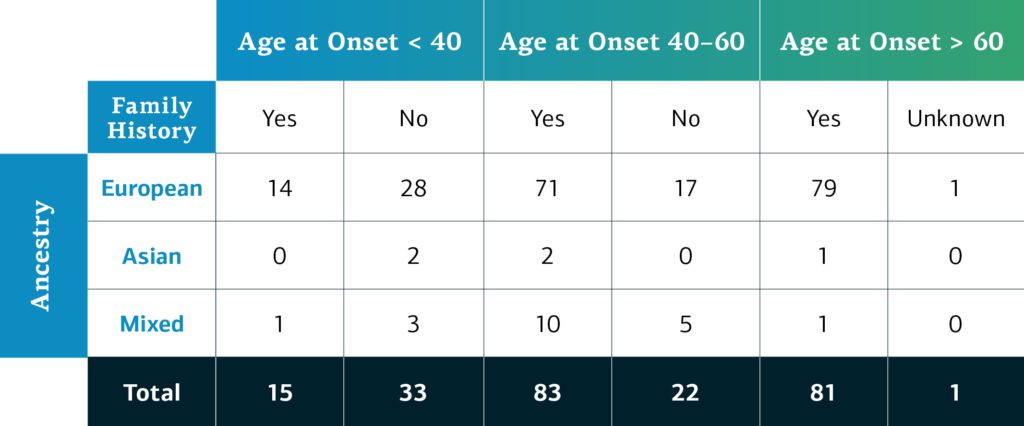
L’ascendance génétiquement déterminée des individus qui participent au réseau GP2 et sont atteints de maladies monogéniques est, pour lors, répartie en quatre grands groupes, car il s’agit des premières données disponibles du réseau monogénique. Au fur et à mesure du traitement des données, d’autres échantillons plus diversifiés deviendront disponibles. Le tableau ci-dessous détaille l’ascendance génétiquement déterminée des participants aux maladies monogéniques dans cette deuxième diffusion du GP2, classés par âge d’apparition et antécédents familiaux.
Les futures diffusions de données permettront d’accroître la diversité des participants. Vous pouvez consulter notre tableau de bord pour suivre nos progrès.
Outre les premières données de séquençage du génome entier, nous avons également inclus un nouveau type de données : les probabilités de variabilité du nombre de copies (CNV) qui exigent que tous les échantillons génotypés aient passé le contrôle de qualité (au niveau du gène plus 250kb de régions flanquantes). Le CNV fait référence à la variation du nombre de répétitions d’un segment d’ADN. Cette variation peut provenir de délétions, d’insertions ou d’autres événements et peut potentiellement fournir plus d’informations sur la façon dont la variation structurelle influence le risque de maladie. L’outil utilisé pour produire les appels probabilistes de CNV est disponible sur le Github du GP2. Il s’agit d’un travail sur le long cours qui s’améliorera au fur et à mesure que nous inclurons des données supplémentaires et que nous ajusterons l’outil. Les notes d’utilisation seront incluses dans le blog sur la première diffusion stable qui est prévue pour le trimestre prochain.
Cette version contient les données du séquençage du génome entier du réseau des maladies monogéniques en plus des données sur les maladies complexes génotypées par le réseau NeuroBooster. De plus amples informations sur la structure du génotype des maladies complexes et des données cliniques sont disponibles dans le blog « The Components of GP2’s First Data Release » ainsi que dans le README, qui est mis à jour à chaque version et est disponible sur les espaces de travail officiels du GP2 Terra. Les données du séquençage du génome entier sur la maladie de Parkinson monogénique sont également détaillées dans le fichier README.
Nous sommes ravis de mettre cette version bêta à la disposition de la communauté de chercheurs sur la maladie de Parkinson et prévoyons bien d’autres choses encore, bientôt disponibles !