En nuestro último artículo de blog, presentamos una introducción a los GWAS con fórmulas estadísticas, flujos de trabajo y ejemplos de los GWAS más recientes sobre la enfermedad de Parkinson. Ahora queremos sugerirle algunas ideas y consejos para que lleve a cabo su propio GWAS.
Control de calidad y procesos de imputación
- Genotipos de alta calidad (no palindrómicos, es decir, sin A/T, G/C, a ser posible, y con una alta puntuación GenTrain de Illumina, indicadora de la calidad del clúster) con una baja tasa de ausencias (preferentemente < 1% por muestra y por SNP).
- Por lo general, céntrese en frecuencias menores y comunes del alelo > 1%, y cruce los dedos para que los grupos de genotipos tengan buen aspecto (deberá inspeccionarlos).
- Compruebe si las muestras presentan altos índices de heterocigosidad, que es un posible signo de contaminación. Una tasa baja de heterocigosidad también podría ser problemática.
- Cuidado con las muestras duplicadas y las muestras crípticamente relacionadas.
- Determine y analice algorítmicamente las muestras por ascendencia genética (nosotros utilizamos una combinación de fastStructure y flashPCA). Es importante maximizar la diversidad, pero hay que tener cuidado con la subestructura de la población y sus efectos en los resultados de los análisis.
- Fíjese en casos en los que la ausencia de SNP no sea una casualidad, por ejemplo en casos de sesgo en que dicha ausencia sea fruto de un estado de control de caso o en haplotipos.
- Puede aplicar múltiples paneles de referencia de imputación para una única población y luego usar el genotipo resultante con la métrica de mejor calidad. Esto es especialmente útil en poblaciones de muestras mezcladas.
Consejos sobre los análisis a nivel de estudio
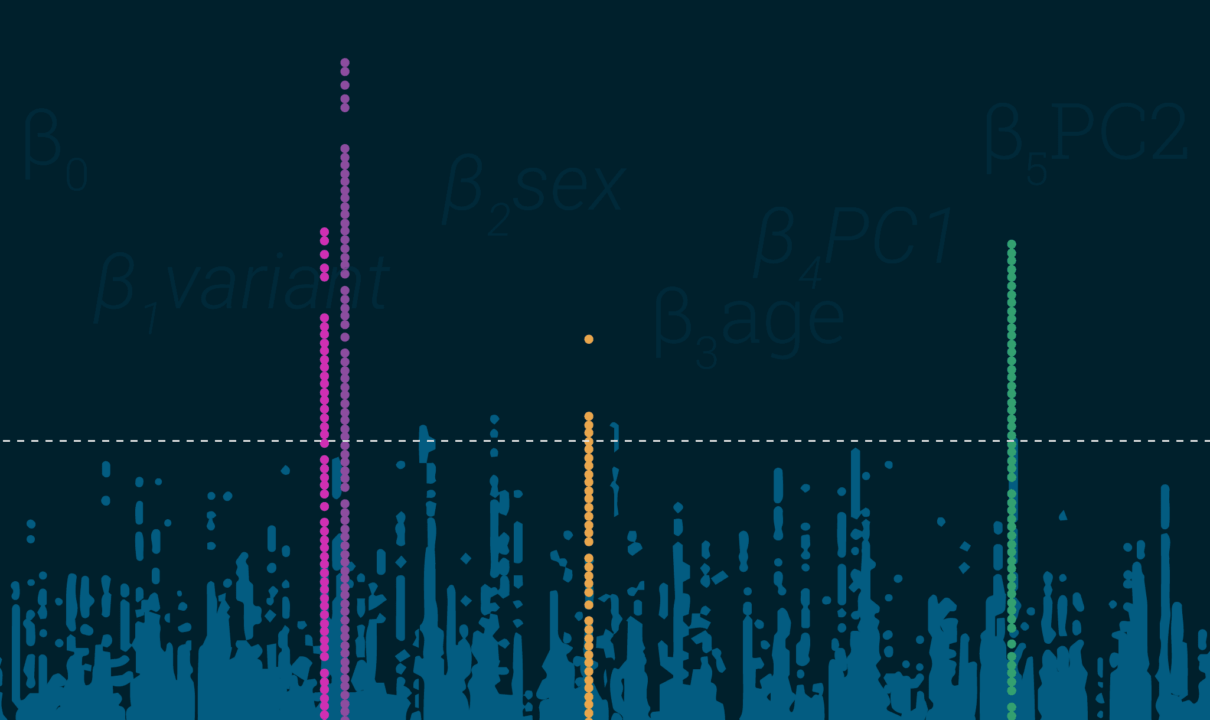
- Regresión. Los GWAS suelen utilizar modelos de regresión lineal para los resultados continuos y de regresión logística para los resultados discretos. Estos cuantifican el riesgo en un SNP al tiempo que consideran las covariables.
- Se pueden utilizar distintos paquetes de software para ejecutar modelos lineales mixtos. Estos modelos dan cuenta de las relaciones con precisión y procesan a escala fina la subestructura de la población.
- PC. El cálculo de las cargas de los componentes principales a partir de los datos de genotipado de todo el genoma brinda un conjunto de covariables que resultará útil para sus modelos de regresión, que a efectos prácticos le permitirá dar cuenta de la subestructura de la población.
Ahora fijémonos en el metanálisis
- Fijo vs aleatorio. Son dos métodos de metanálisis que permiten combinar datos para elaborar síntesis estadísticas a partir de silos de datos o publicaciones. Los metanálisis de efecto fijo suelen ser útiles para los análisis de descubrimiento y, en general, son más potentes, aunque algunos preferimos los modelos de efecto aleatorio, ya que brindan análisis más conservadores y tienen en cuenta la heterogeneidad entre los silos de datos.
- Estimaciones de la heterogeneidad. A menudo expresada como Q de Cochrane o I2. Son fundamentales porque dan una idea tanto del sesgo por efecto de los valores atípicos como del potencial generalizable de los resultados.
Resultados del escrutinio
- Lambdas a nivel del estudio. Deben mantenerse entre 0.95 y 1.05, cualquier otro valor supondría un problema.
- Lambda total y lambda 1000. El cálculo de lambdas tanto a nivel de estudio como de metanálisis puede ser complicado cuando existen desequilibrios entre casos y controles. Un gran exceso de controles puede inflar artificialmente la estadística lambda. En este caso, utilice lambda1000, a la que se aplica una escala de 1000 casos y 1000 controles. Una vez más, encontrará el código correspondiente en nuestro repositorio de GitHub.
- Intercepción de la puntuación de desequilibrio del ligamiento (LD). Alternativa / complemento a lambda y más robusto que la estructura de LD y caso: desequilibrio de control.
- El LD alcanza los valores más altos en segmentos que presentan uniformidad. Esto es bastante sencillo: al representar sus resultados, los segmentos deben parecer los rascacielos de Manhattan (o de la ciudad que prefiera, lo siento D.C.), y no una tormenta de nieve.
- Replicación. Siempre es buena, pero a veces no hay conjuntos de datos disponibles. Cuando no se disponga de datos adicionales, intente una combinación de metanálisis con exclusión y validación cruzada.
Algunos análisis post-GWAS
- O «qué hacer una vez completados sus metanálisis GWAS». Tenga en cuenta que el GP2 publicará más artículos de blog y cursos sobre estos temas.
- Análisis condicionales. Use el SNP más significativo en una región como covariable y vuelva a ejecutar los análisis o use una herramienta como GCTA. Puede obtener varias señales independientes por locus.
- Mapeo fino. Se puede hacer mediante el análisis factorial bayesiano aproximado en paquetes como coloc, incluyendo la colocalización bayesiana (aprovechando datos genómicos de referencia para la expresión de genes o parámetros similares).
- Peso de predicciones y TWAS. Existen diversos paquetes para aprovechar el peso externo de los estudios de expresión génica, metilación o cromatina, etc. para identificar los genes putativos conectados mecánicamente que se esconden en sus datos GWAS.
- En cuanto a predicciones creadas a propósito basadas en aprendizaje automático (AA), nuestro paquete favorito es GenoML, porque está básicamente automatizado y facilita el AA en el campo de la genómica.
Para más información, encontrará nuestro código y pipelines en el GitHub del GP2.
Este blog es obra conjunta de Hampton Leonard, Mike Nalls, Yeajin Song y Dan Vitale. Visite la página del Grupo de Trabajo de Análisis de Datos sobre Enfermedad de Parkinson de Etiología Compleja del GP2 para obtener más información.