
Dans notre dernier blog, nous avons présenté les GWAS (études d’association pangénomique) avec les formules statistiques, les flux de travail et les exemples de la dernière étude d’association pangénomique consacrée à la maladie de Parkinson. Cette fois-ci, nous souhaitons vous donner quelques notions et conseils supplémentaires pour lancer votre propre étude pangénomique.
Contrôle de qualité et processus d’imputation
- Génotypes de haute qualité (non-palindrome, c-à-d non A/T, G/C si possible, avec un score Illumina élevé indiquant la qualité du groupe) avec peu de lacunes (<% par échantillon et par PSN, de préférence).
- De façon générale, concentrez-vous sur les fréquences communes d’allèles mineurs >1%. S’ils sont inférieurs, espérez que les groupes de génotype soient prometteurs (vous devrez les vérifier).
- Vérifiez si les échantillons ont un fort taux d’hétérozygotie car cela indique une contamination potentielle. Un taux d’hétérozygotie feu élevé peut également s’avérer problématique.
- Faites attention aux échantillons en dupliqué et aux échantillons qui leur sont mystérieusement associés.
- Vérifiez et passez au crible les échantillons avec l’algorithme en fonction de l’ascendance génétique (nous utilisons une combinaison de fastStructure et de flashPCA). Tachez de maximiser la diversité mais, faites attention aux sous-structures de populations et à leur incidence sur les résultats des analyses.
- Cherchez les PSN délibérément manquants, y compris en raison d’absence par cas : contrôle de statut ou haplotype.
- Rien ne vous empêche d’utiliser des panels de référence à imputation multiple sur une même population et utilisez le génotype obtenu avec la meilleure qualité métrique. C’est particulièrement utile pour les échantillons de populations mélangées.
Conseils pour les analyses d’études
- Régression – Les études d’association pangénomique utilisent généralement des modèles de régression pour les résultats en continu et la logistique pour les résultats ponctuels. Cela permet de quantifier le risque au niveau du PSN tout en tenant compte des co-variables.
- Divers paquets de logiciels peuvent être utilisés pour opérer des modèles linéaires mixtes, lesquels sont fiables pour la préparation et l’étalonnage précis des sous-structures de population.
- PC – Calculer les principaux chargements de composant à partir du génotypage des données pangénomiques permet d’obtenir un ensemble de co-variables utiles pour vos modèles de régression, vous permettant réellement de compter sur une sous-structure de population.
Maintenant, passons à la méta-analyse
- Fixe ou aléatoire – Il s’agit de méthodes de méta-analyse qui permettent de combiner des données au niveau du résumé statistique, de façon transversale aux silos et/ou publications. Les méta-analyses sont souvent utiles pour les analyses de découverte et généralement plus puissantes, bien que certains parmi nous préfèrent les modèles à effets randomisés, plus conservateurs et qui tiennent compte de l’hétérogénéité des silos de données.
- Estimations de l’hétérogénéité – Souvent appelées test Q de Cochran ou I2, elles sont très importantes pour avoir une idée de l’écart des valeurs aberrantes.
Examen de contrôle
- Niveau d’étude des lambdas – les maintenir dans une fourchette de 0,95 à 1,05 ; toute autre valeur posera problème.
- Lambdas généraux et lambda 1000 – Le calcul des lambdas à l’échelle de l’étude ou de la méta-analyse peut être délicat en cas de déséquilibre du contrôle. Un nombre massif de contrôles peut faire gonfler artificiellement les statistiques lambda. Dans ce cas, utilisez le lambda 1000 car il est établi pour 1000 cas et 1000 contrôles. Nous vous rappelons que ce code se trouve dans notre recueil GitHub.
- Interception du score par déséquilibre de liaison (DL) – Méthode alternative ou complémentaire à la méthode lambda et plus solide pour la structure et le cas DL : contrôler les déséquilibres.
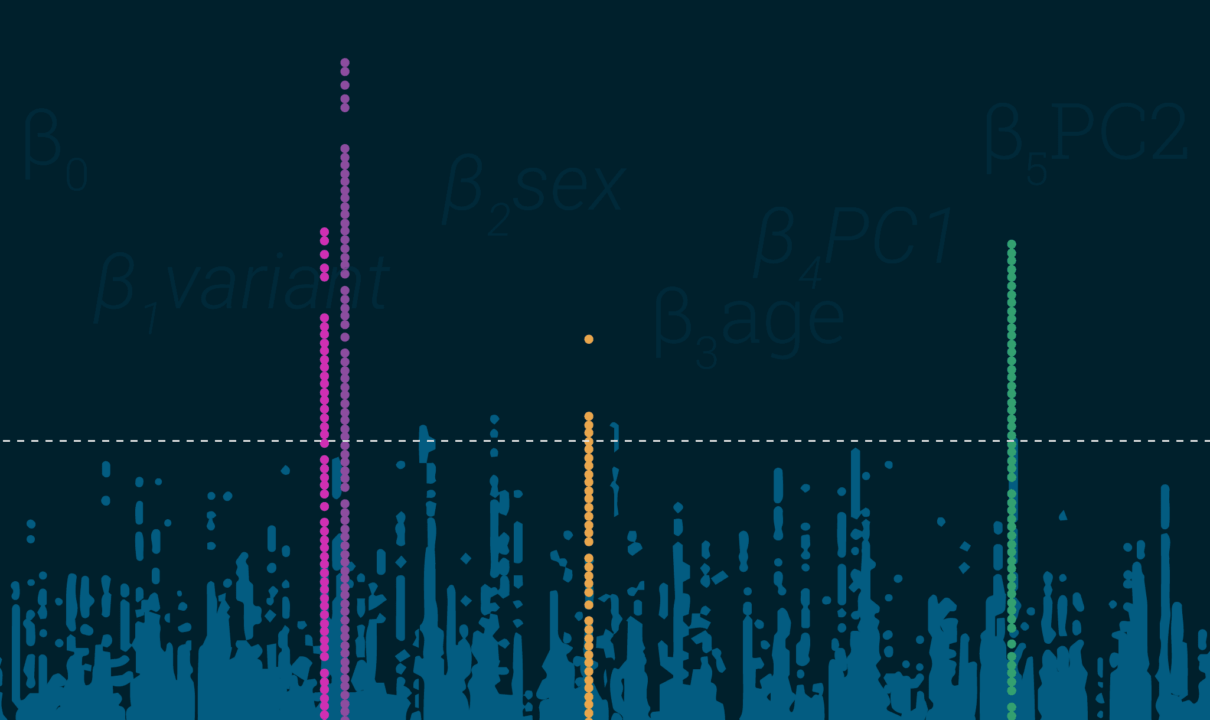
- Le DL atteint son maximum sur des zones affichant une uniformité. C’est assez simple car lorsque regroupés en zones, vos résultats devraient ressembler aux tours de Manhattan (ou de la ville de votre choix) et pas à une tempête de neige.
- Réplication – C’est toujours bien mais parfois, vous n’avez pas les ensembles de données à disposition. Lorsque les données supplémentaires ne sont pas disponibles, essayez un mélange entre le fait de laisser une méta-analyse de côté et la validation croisée.
Quelques analyses post-études d’association pangénomique
- ou « que faire une fois que vous avez terminé votre analyse de l’étude d’association pangénomique ». Notez que le GP2 proposera d’autres blogs et formations sur ce sujet.
- Analyses conditionnelles – Utilisez le PSN le plus pertinent dans une région donnée comme co-variable et relancez les analyses ou utilisez un outil tel que le GCTA. Il se peut que vous ayez des signaux indépendants multiples par locus.
- Cartographie fine – Elle peut être obtenue par le biais de l’analyse de facteur Bayes en lot tels que coloc incluant la colocalisation bayésienne (utilisation des données de référence génomique pour l’expression génique ou une métrique similaire).
- Poids de prédiction et TWAS – un ensemble de paquets existent pour exploiter la charge extérieure de l’expression génique, méthylation ou études de chromatine, etc. afin d’identifier les éventuels gènes reliés mécaniquement se cachant dans vos données d’étude d’association pangénomique.
- ML – conçu pour les projections, notre paquet favori est GenoML parce qu’il est essentiellement automatisé et rend facile l’apprentissage machine (AM) en génomique.
Pour plus d’informations, vous trouverez notre code et lignes sur le GitHub du GP2.
Ce blog a été co-écrit par Hampton Leonard, Mike Nalls, Yeajin Song et Dan Vitale. Veuillez consulter la page du GP2 du groupe de travail sur l’analyse des données – Maladie complexe pour en savoir plus sur les auteurs.


