Comprendre les études d’association pangénomique… ou que faire de tous ces petits taux d’incidence ? Ce qui suit est une grossière simplification de ce que sont les études d’association pangénomique, et pourra être empreint d’une ironie bienveillante.
Qu’est-ce que les études d’association pangénomique (GWAS) peuvent-elles nous dire de la maladie ? Pourquoi tester plus d’un million de caractéristiques indépendantes du génome et subir un abus notoire de test à répétition ? La réponse est assez facile : pour deux raisons toutes simples. Premièrement, chaque région de gènes associée au candidat (le locus) élargit la portée de nos connaissance sur la maladie. Deuxièmement, tous ces petits effets cumulés observés, chacun apportant une hausse marginale de risque génétique, nous aide à mieux prédire le risque et à préciser les critères de diagnostique. On dit que les études d’association pangénomique sont dénuées d’hypothèses mais en réalité, l’hypothèse est un « variant ou un ensemble de variants du génome qui peut être un trouble-fête, c’est pourquoi, nous devons creuser la question. »
A un niveau plus élémentaire, une étude d’association pangénomique n’est qu’un ensemble d’équations, chacune s’intéressant à un variant génomique à la fois. Voyons comment cela s’applique à la maladie de Parkinson.
Maladie de Parkinson (1 pour oui, 0 pour non) ~ β0 + β1variant + β2sexe + β3âge + β4PC1 + β5PC2
Si vous êtes habitué aux statistiques, votre formule GWAS classique n’est qu’une simple formule à régression multiple. Cette équation spécifique teste s’il est possible de prévoir qu’une personne est atteinte de la maladie de Parkinson en vérifiant la présence d’un variant spécifique dans le génome de cette personne. Outre le variant testé, nous incluons d’autres facteurs tels que le sexe, l’âge et les principaux éléments qui expliquent le niveau individuel de l’ascendance génétique.
Voyons maintenant les taux d’incidence et les bêta. Dans l’équation précédente, le terme « βn » devant les variables correspond aux coefficients. Ils nous renseignent sur les probabilités ou la prédisposition à la hausse ou à la baisse d’être atteint de la maladie de Parkinson, en fonction de la présence de cette variable. Dans les manuscrits GWAS, vous verrez que cet effet de variants est repris comme étant la valeur bêta ou le taux d’incidence. Voyons un exemple pour interpréter les deux cas. Dans l’étude la plus récente et à plus grande échelle réalisée à ce jour sur la maladie de Parkinson (Nalls 2019), l’un des nouveaux variants qui semble être associé à la maladie de Parkinson est le variant rs76116224 sur le chromosome 2. La valeur bêta associée à ce variant est de 0,110. La taux d’incidence est de 1,12 (intervalle de confiance : 1,08–1,16). La valeur bêta représente par unité la hausse ou la baisse du résultat. Donc, dans cette étude :
- Une copie du variant rs76116224 représente une augmentation du risque estimé de la maladie de Parkinson de 0,110 * 1 = 0,110.
- Deux copies du variant rs76116224 correspondent à une augmentation du risque estimé de la maladie de Parkinson de 0,110 * 2 = 0,220.
Les taux d’incidence (RI) sont les exposants des coefficients bêta mais leur interprétation est légèrement différente. Un RI supérieur à 1 est associé à une plus grande probabilité du résultat, un RI de 1 signifie pas d’association et un RI inférieur à 1 signifie une probabilité moindre. Donc un RI de 1,12 signifie qu’il faut s’attendre à une hausse de 12 % des chances d’avoir la maladie de Parkinson pour chaque unité d’augmentation de la copie du variant.
Maintenant que vous avez l’équation, vous n’avez qu’à effectuer le calcul pour chaque variant de vos données. Donc, selon la façon dont vos données ont été traitées, cela fait environ 25 375 550 ! Heureusement nous avons de gentils logiciels qui se chargent de faire les calculs à notre place.
Généralement, chaque étude est menée dans un centre à part, à partir de leurs propres données, par ascendance et cohorte individuelle. Toutefois, plus nous incluons de personnes, plus grandes sont les chances de détecter des variants aux effets rares. Pour augmenter les chiffres, chaque étude est combinée dans le cadre d’une méta-analyse. Les sites collaboratifs se mettent d’accord en amont sur la définition de la maladie ou du résultat, quel code ou infrastructure informatisée utiliser et dans certains cas, le nom des auteurs sur les documents produits à l’issue de l’étude. Chaque site d’étude met généralement en place des paramètres simples de contrôle de qualité pour la gamme de données génotypique de l’étude en question, ainsi que sur une méthode d’imputation (qui utilise le même panel de référence pour les variants génétiques non testés). Les paramètres de contrôle de qualité choisis doivent avoir été testés avant le lancement de l’étude d’association pangénomique.
Une fois le contrôle de qualité effectué et le lancement de l’étude pour chaque personne, des techniques de méta-analyses à partir de statistiques élémentaires sont utilisées pour combiner les statistiques résumées de l’étude pangénomique dans un seul ensemble de résultats compilés par variant. A partir de là, après d’autres contrôles de qualité, vous pouvez commencer à chercher les variants ayant une valeur p < 5e-08 (ce raccourci est généralement choisi dans les études pangénomiques pour la correction de tests multiples, par millions, mais il est préférable d’être plus rigoureux encore). Les variants qui passent le seuil significatif sont considérés comme étant associés de façon significative au résultat testé, mais il est important d’essayer de reproduire ces résultats dans des ensembles de données supplémentaires.
Les variants découverts par les études pangénomiques nous donnent les cibles à étudier et nous aident à améliorer notre connaissance de la maladie. Chaque nouvelle cible nous aide à renforcer notre capacité à prévoir les chances d’une personne de développer un résultat donné. Ces informations seront nécessaires alors que nous nous dirigeons vers une médecine de précision dans le traitement de la maladie. Avec le GP2, nous sommes décidés à élargir les connaissances sur la maladie de Parkinson. Nombre de processus utilisés par les études pangénomiques sont automatisés et le code que nous utilisons est public, transparent et en croissance constante. Nous mettons l’accent sur la diversité c’est pourquoi nous traitons des analyses intégrées sur plusieurs silos de données, analysant des cohortes harmonisées, unifiées si possible et procédant à des méta-analyses, le cas échéant.
La création de flux de travail uniformisés et clairs et d’un code, dont nous espérons qu’il sera utilisé par une multitude de chercheurs et d’institutions dans le monde entier, est le fruit d’un effort considérable. Notre objectif est de faire du GP2 le projet d’analyse de science ouverte le plus transparent au monde.
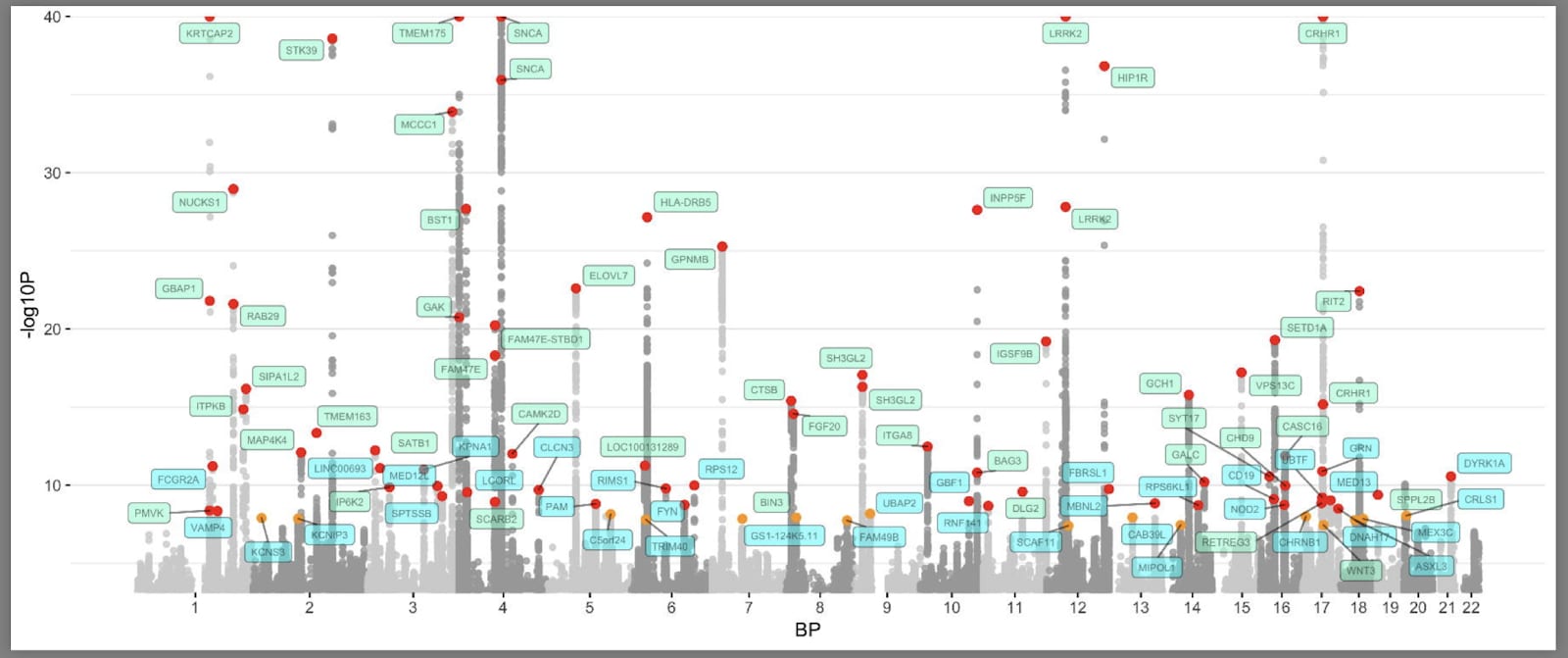
Voici à quoi ressemble le «complot de Manhattan» (baptisé comme les tours de Manhattan) pour l’étude pangénomique sur la maladie de Parkinson (Nalls et. al., 2019) Vous trouverez le code à cet effet ici.
Vous trouverez notre code et les pipelines au GitHub du GP2 et surveillez le post du blog de suivi qui donne des conseils et recommandations pour le lancement de votre propre étude pangénomique.