
¿De qué va eso de la razón de momios? A continuación presentamos una simplificación excesiva de los estudios GWAS, acompañada, por qué no, de una dosis de sarcasmo.
¿Qué nos cuentan los estudios de asociaciones de genoma completo (GWAS, por sus siglas en inglés) acerca de las enfermedades? ¿De qué sirve hacer ensayos con más de un millón de características independientes del genoma y soportar el abuso de la infinidad de ensayos? La respuesta no tiene ningún secreto: por dos razones muy simples. En primer lugar, cada región candidata asociada a los genes (también conocida como locus) nos permite ampliar el alcance de nuestros conocimientos sobre una enfermedad. En segundo lugar, todos esos pequeños efectos acumulativos que se observan, cada uno de los cuales supone un aumento marginal del riesgo genético, nos ayudan a predecir mejor el riesgo y a afinar los criterios de diagnóstico. Se dice que los GWAS no parten de ninguna hipótesis, pero en realidad sí parten de una, a saber: «que una variante o conjunto de variantes del genoma podría tener efectos nocivos…. tenemos que investigarlo más a fondo».
A un nivel muy básico, un GWAS no es más que una serie de ecuaciones, cada una de las cuales examina una variante genómica. Desglosemos esta definición aplicándola a la enfermedad de Parkinson.
Enfermedad de Parkinson (1 si sí, 0 si no) ~ β0 + β1variante + β2sexo + β3edad + β4PC1 + β5PC2
Si tiene nociones de estadística, habrá observado que la fórmula estándar de un GWAS corresponde a una simple fórmula de regresión múltiple. Esta ecuación en particular pone a prueba si podemos predecir si una persona tiene la enfermedad de Parkinson o no a partir de la presencia de una variante específica en el genoma de esa persona. Además de la variante analizada, incluimos otros factores como el sexo, la edad y los componentes principales que explican la herencia genética a nivel individual.
Ahora enfoquémonos en la razón de momios y las betas. En la ecuación anterior, los términos «βn» delante de las variables corresponden a coeficientes. Estos nos indican cuánto aumenta o disminuye la probabilidad estimada o el «riesgo» de padecer la enfermedad de Parkinson en función de la presencia de esa variable. En los manuscritos de los GWAS, estos tamaños de efecto de las variantes se presentan como valores beta o razón de momios. Veamos un ejemplo para interpretar ambos. En el GWAS sobre EP más reciente y más amplio hasta la fecha (Nalls 2019), una de las nuevas variantes que se ha asociado significativamente a la EP es la variante rs76116224 en el cromosoma 2. El valor beta asociado a esta variante es de 0.110. La razón de momios es de 1.12 (intervalo de confianza: 1.08–1.16). El valor beta hace referencia al aumento o disminución por unidad del resultado. Así que, en este estudio:
- Una copia de la variante rs76116224 supone un aumento estimado del riesgo de contraer la enfermedad de Parkinson de 0.110 * 1 = 0.110.
- Dos copias de la variante rs76116224 suponen un aumento estimado del riesgo de contraer la enfermedad de Parkinson de 0.110 * 2 = 0.220.
La razón de momios es el exponente de los coeficientes beta, pero se interpreta de forma ligeramente distinta. Una razón de momios superior a 1 se asocia a una mayor probabilidad del resultado, una razón de momios igual a 1 significa que no hay asociación, y una razón de momios inferior a 1 se asocia a una menor probabilidad del resultado. Así, una razón de momios de 1.12 significa que esperaremos ver un aumento del 12 % en las probabilidades de tener la enfermedad de Parkinson por un aumento unitario en la copia de la variante.
Ahora que ya sabe la ecuación, solo tiene que calcularla para cada variante de sus datos. Así que, dependiendo de cómo se hayan procesado sus datos, ¡solo tendrá que repetir el cálculo unas 25,375,550 veces! Por suerte, tenemos un buen software que ejecuta todas estas ecuaciones por nosotros.
Típicamente, cada GWAS se realiza en un centro separado y con datos propios, por ascendencia genética y por cohorte individual. Sin embargo, cuantas más personas incluyamos, mayores serán las posibilidades de detectar variantes de efectos poco frecuentes. Para aumentar estas cifras, cada GWAS se combinará en un metanálisis. Los centros de colaboración acordarán de antemano cómo se define la enfermedad u otro resultado, qué código e infraestructura informática se utilizará y, en algunos casos, incluso el orden de autoría de los trabajos resultantes. Cada centro de estudio suele acordar unos parámetros básicos de control de calidad (CC) para sus datos de matriz de genotipado a nivel del estudio, así como un método de imputación consensuado (utilizando el mismo panel de referencia para inferir las variantes genéticas no analizadas). Se deberán verificar los parámetros de control de calidad establecidos antes de ejecutar cada GWAS.
Una vez realizado el control de calidad y ejecutado cada GWAS individual, pueden utilizarse técnicas de metanálisis de estadística básica para combinar las estadísticas resumidas de los GWAS en un único conjunto de resultados agregados por variante. A partir de aquí, y tras algún control de calidad adicional, se pueden empezar a buscar variantes con valores p < 5e-08 (este es el umbral que suele establecerse en los GWAS para corregir millones de ensayos, pero siempre es recomendable establecer un umbral más estricto). Las variantes que superan este umbral se consideran significativamente asociadas con el resultado sometido a prueba, pero es importante intentar replicar los resultados en conjuntos de datos adicionales.
Las variantes identificadas por los GWAS nos proporcionan objetivos de investigación y ayudan a profundizar nuestro conocimiento de las enfermedades. Cada nuevo objetivo de investigación nos ayuda a aumentar nuestra capacidad de predecir con precisión el riesgo de que una persona exhiba un resultado determinado. Estos conocimientos serán necesarios a medida que avancemos hacia un enfoque de medicina de precisión en el tratamiento de las enfermedades. Desde el GP2, nos comprometemos a ampliar este conocimiento en torno a la enfermedad de Parkinson. Muchos de nuestros procesos de GWAS están automatizados, y el código que utilizamos es público, transparente y se amplía constantemente. Nos enfocamos en la diversidad, por lo que llevamos a cabo análisis integrados entre silos globales de datos, analizamos cohortes armonizadas y unidas cuando es posible, pero también llevamos a cabo metanálisis cuando procede.
Nos esmeramos para construir flujos de trabajo y códigos uniformes y claros, que esperamos que sean de utilidad para investigadores e instituciones de todo el mundo. Nuestro objetivo es hacer de GP2 el proyecto de análisis basado en la ciencia accesible más transparente del planeta.
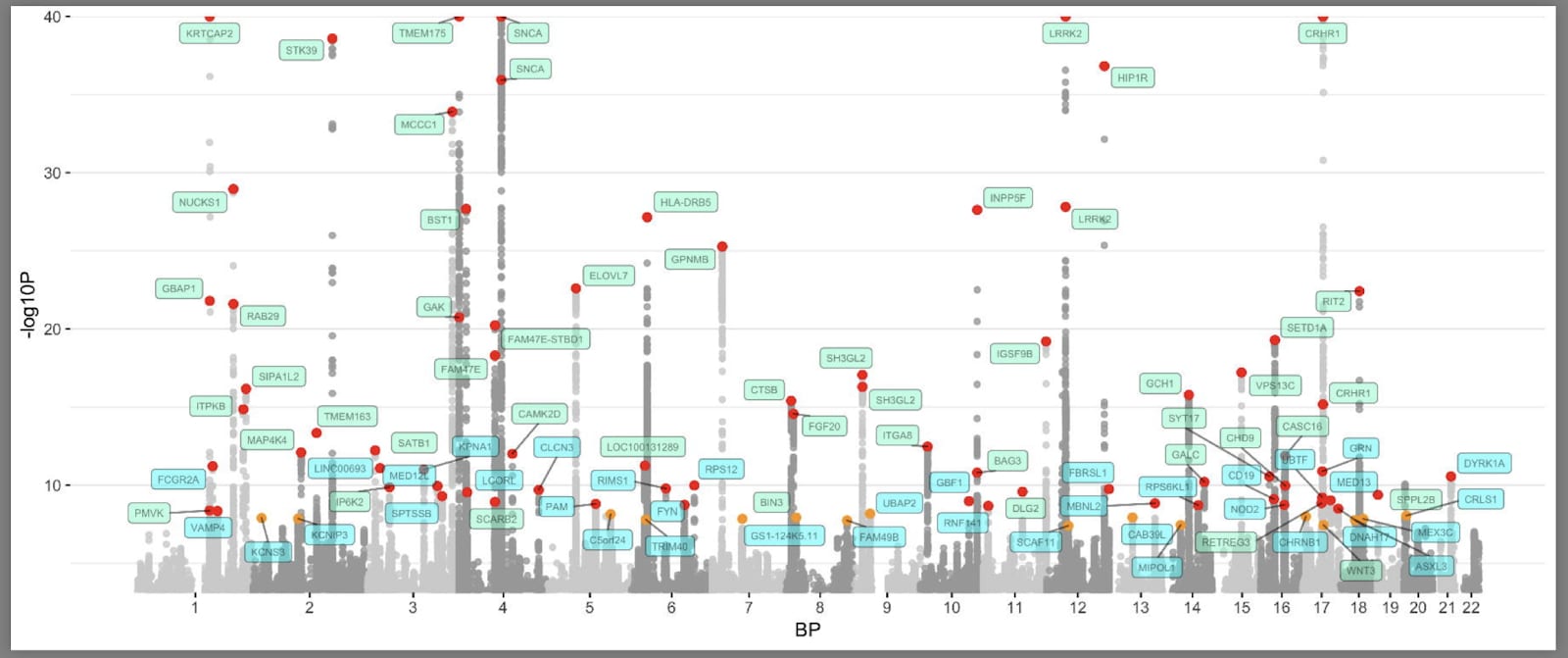
Este es el «diagrama de Manhattan» (debe su nombre a las torres de Manhattan) para el GWAS de la enfermedad de Parkinson (Nalls et al., 2019). Encontrará el código utilizado aquí.
Encontrará nuestro código y nuestros procesos en GP2 GitHub. No se pierda nuestro próximo artículo de blog, donde le daremos ideas y consejos para que ejecute su propio GWAS.



