
了解GWA……或所有这些小几率是什么?以下对GWAS研究的解释是过度简化的版本,其中可能包括善意的讽刺。
全基因组关联研究(GWAS)能告诉我们有关疾病的什么?为什么要测试全基因组一百多万个独立的特征,并忍受严重的多重测试的滥用?答案很简单,有两个简单原因。首先,每个候选的相关基因区域(也称为基因座)扩大了我们对疾病的了解范围。其次,您观察到的所有累积的小影响,每一个都会使遗传风险稍有增加,有助于我们更好地预测风险并完善诊断标准。据说GWAS是没有假设的,但实际上其假设是“基因组中的一个变体或一组变体可能正在做一些不好的事情,我们需要了解更多”。
在最基本的层面上,GWAS只是一系列方程式,每个方程式一次查看一个基因组变体。下面我们在帕金森病的场景里来分解一下。
帕金森病(是1,否0) ~ β0 + β1变体 β2性别+ β3年龄+ β4PC1 + β5PC2
如果您熟悉统计信息,那您标准的GWAS公式就只是一个简单的多元回归公式。这个特定的方程式测试我们是否可以使用某人基因组中的特定变体来预测某人是否患有帕金森病。除了测试的变体之外,我们还包括其它因素,例如性别、年龄和解释个体水平遗传祖先的主要成分。
现在我们来讨论几率和贝塔值。在上式中,变体前面的‘βn’项是系数。这些系数告诉我们,该变量的存在估计让帕金森病的可能性或“风险”增加或减少了多少。在GWAS手稿中,您会看到这些变体的效应大小被称为贝塔值或几率。举个例子来阐释这二者。在迄今为止最新、最大规模的PD GWAS中(Nalls 2019),发现与PD显著相关的新变体之一是2号染色体上的rs76116224变体。这个变体相关的贝塔值是 0.110。几率是1.12(置信区间:1.08–1.16)。贝塔值是结果中每单位的增加或减少。因此,在这个研究中:
- 一份rs76116224变体的副本估计使帕金森病风险增加了0.110 * 1 = 0.110。
- 两份rs76116224变体的副本估计使帕金森病风险增加了0.110 * 2 = 0.220。
几率(OR)是贝塔系数的指数,但解释稍有不同。大于1的OR表示结果的几率较高,OR等于1表示没有关联,小于1的OR标识结果的几率较低。因此,OR为1.12意味着变种副本每增加一个单位,患帕金森病的几率预计将增加12%。
既然有了公式,就可以计算数据中每一个变体的相关数值了。取决于您怎么处理数据,也就计算25,375,550次吧!好在我们有很好的软件来帮我们运算。
通常,每个GWAS都是在单独的中心根据各自的祖先和每个队列的数据来进行计算的。但是,我们包括的人员越多,发现罕见效应变异的机会就越大。为了增加这些数量,每个GWAS都将进行荟萃分析。协作研究场所将提前就疾病或其它结果的定义、使用的代码和计算基础结构、在某些情况下甚至在结果论文的作者顺序上达成一致。每个研究地点通常都同意其研究水平的基因分型阵列数据的基本质量控制(QC)参数,以及估算的共识方法(使用同一参考面板来推断未测定的遗传变异)。必须在运行GWAS之前执行所选的质量控制参数。
完成质量控制并运行每个单独的GWAS后,将使用来自基本统计信息的荟萃分析技术将GWAS摘要统计信息组合为每个变体的单个汇总结果集。从这里开始,经过一些额外的质量控制,您就可以开始寻找p值<5e-08的变体(通常为GWAS选择此临界值以校正数百万的多次测试,但最好是更加严格)。通过此显著性阈值的变体被认为与测试结果显著相关,但重要的是尝试在其它数据集中复制这些发现。
GWAS发现的变体为我们提供了研究的目标,帮助增进我们对疾病的了解。每一个新的目标都可以帮助我们建立能力,以准确地预测某人发展特定结果的风险。当我们朝着治疗疾病的精确医学方法迈进时,这些见解将非常必要。我们致力于跟GP2一起扩大对帕金森病的了解。我们很多GWAS的程序都实现了自动化,我们使用的代码都是公开的、透明的,并在不断地增加。我们关注多样性,因此我们在全球数据孤岛中进行综合分析,在可能的情况下进行协调统一的队列分析,并在适当时进行荟萃分析。
我们构建统一、清晰的工作流和代码方面付出了巨大的努力,希望可为世界各地的多个研究机构和人员所用。我们的目标是将GP2打造成世界上最透明的开放科学分析项目。
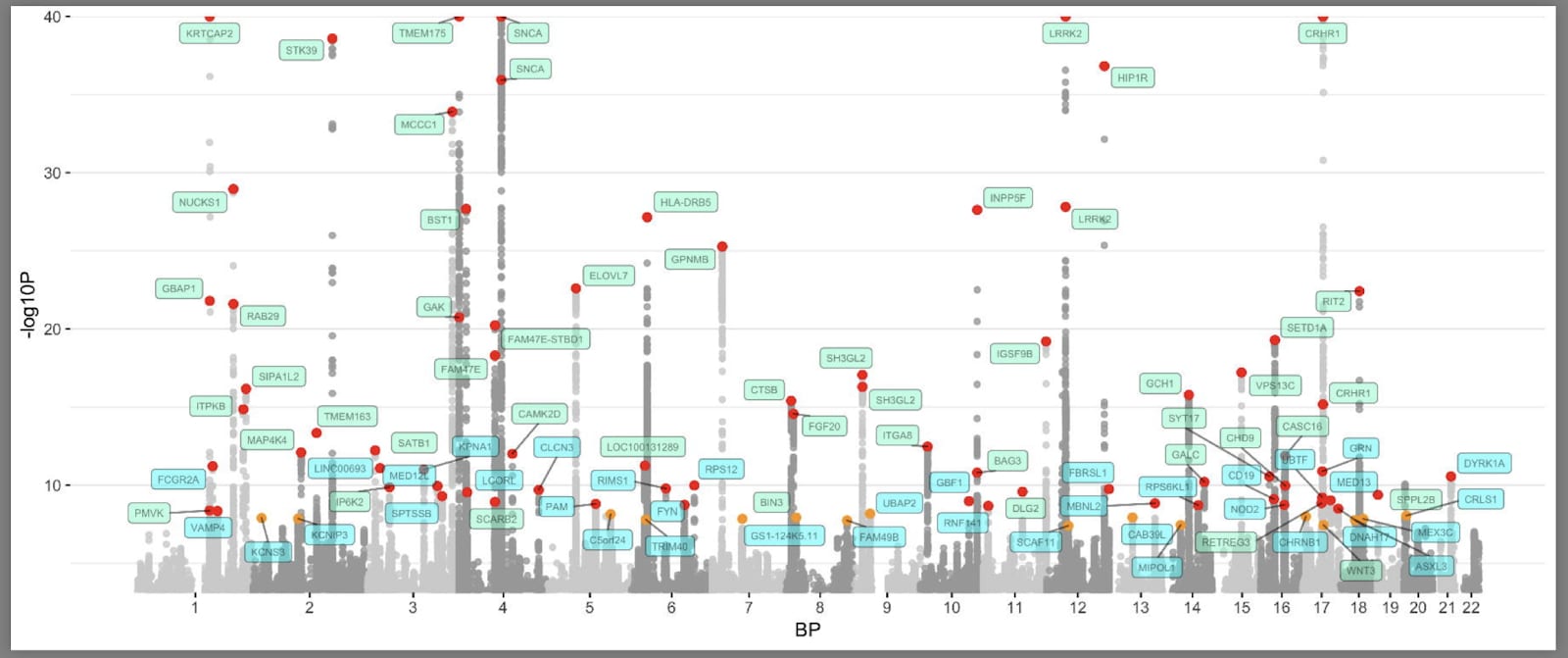
针对帕金森病GWAS的“曼哈顿图”(以曼哈顿的塔楼命名)现在是这样(Nalls等人,2019)。代码可在 这里找到。
您可以在GP2 GitHub上找到我们的代码和后续工作,并查找后续博客文章,其中提供了有关运行自己的GWAS的见解和提示。



