La enfermedad de Parkinson (EP) ha sido el foco de una extensa investigación genética. Los investigadores han identificado más de 90 variantes genéticas que pueden afectar al riesgo de que una persona presente EP, lo que revela que se trata de una enfermedad compleja en la cual intervienen muchas vías moleculares.
Sin embargo, la falta de diversidad genética en los estudios sobre la EP, centrados principalmente en poblaciones de ascendencia europea, ha dificultado una comprensión global de la EP. ¿Qué variantes genéticas de la EP tienen más probabilidades de ser causales? ¿Qué variantes podrían ser clínicamente útiles en todas las poblaciones? Para responder a estas preguntas se necesitan más datos sobre la diversidad de ascendencia.
Una colaboración mundial: ascendencia diversa y a gran escala
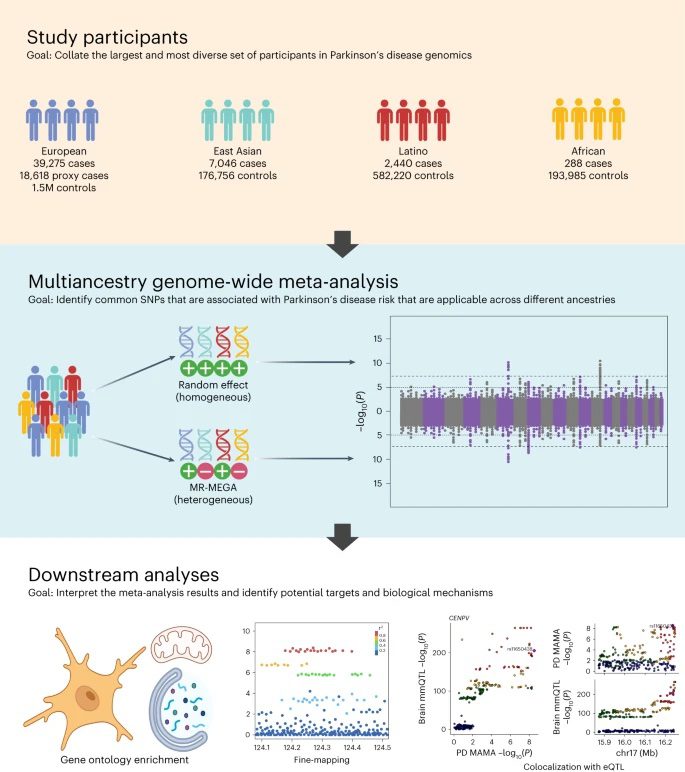
Un nuevo estudio colaborativo publicado en Nature Genetics ofrece una comprensión más inclusiva y global del panorama genético de la EP. Se trata del estudio más diverso y a mayor escala de su clase, gracias en gran parte a la colaboración mundial y al intercambio de datos encabezados por el Global Parkinson’s Genetics Program (GP2). El estudio analiza datos que representan a 49,049 personas con EP, 18,785 personas cuyos padres tienen o tuvieron EP y más de 2 millones de personas neurológicamente sanas de ascendencia europea, asiática oriental, latinoamericana y africana.
Investigadores de los NIH, el GP2, 23andMe y colaboradores de los EE. UU., el Reino Unido, Perú y Singapur seleccionaron estos datos de ascendencias diversas de FinnGen, 23andMe, UK Biobank y otros repositorios de datos de libre acceso. Con estos datos, identificaron 78 regiones del genoma (loci) significativamente asociadas a la EP.
El análisis de datos procedentes de múltiples grupos de ascendencia distintos presenta retos singulares, pero están surgiendo nuevos programas de software y herramientas de análisis que pueden aprovechar datos cada vez más diversos. Gracias a estas herramientas y a las iniciativas de colaboración en todo el mundo, los investigadores pueden ahora identificar regiones genéticas significativamente asociadas al riesgo de la EP en poblaciones de ascendencias múltiples. Estas regiones tienen más probabilidades de contener variantes causales.
Los investigadores del estudio aplicaron un enfoque denominado metaanálisis de asociación de genoma completo (GWAS) de ascendencia múltiple. GWAS es un método que consiste en analizar los genomas de muchas personas, con el fin de identificar variantes de secuencia específicas en regiones genómicas (o loci) que se dan con mayor frecuencia en quienes padecen una enfermedad concreta en comparación con quienes no la padecen. Los investigadores reunieron datos de GWAS publicados en diversos repositorios de datos para sumar fuerza estadística y solidez a la búsqueda de factores de riesgo genéticos de la EP.
Al incluir datos de poblaciones no europeas, los investigadores pudieron identificar muchos loci genéticos novedosos que no se habían observado con anterioridad, cuando solo se analizaban poblaciones de ascendencia europea. Mientras que la mayoría de los 78 loci de EP identificados en este estudio coinciden con regiones ya asociadas a la EP, 12 de ellos son potencialmente nuevos. Parece que los nuevos loci están relacionados con genes que ayudan a descomponer o metabolizar proteínas, controlar la actividad de las mitocondrias y activar respuestas inmunitarias en el cerebro.
La mayoría de los loci identificados se asociaron significativamente al riesgo de EP en todas las ascendencias estudiadas, incluidas las poblaciones europeas. Estos resultados sugieren que todas las poblaciones comparten las numerosas vías que pueden causar EP.
Hacia una medicina personalizada: mapeo genético fino e implicaciones funcionales
Los investigadores también lograron determinar la variante genética causal específica en al menos 6 de los loci identificados, en un proceso denominado mapeo genético fino. El mapeo genético fino estima qué variantes específicas dentro de cada loci son probablemente responsables de conferir riesgo de enfermedad en una población determinada.
«Uno de los problemas del GWAS es que nunca se sabe qué variante específica es la funcional o causal en un locus identificado», explica Ignacio (Nacho) Mata, científico del Learner Research Institute, profesor adjunto de Medicina Molecular en el Learner College of Medicine de Cleveland Clinic y autor del estudio. «En cualquier ‘resultado positivo’ del GWAS o región del genoma significativamente asociada a una enfermedad podría haber cientos de posibles variantes candidatas».
Identificar las variantes funcionales en los loci GWAS significativos y descubrir cómo afectan a la expresión génica es clave para los estudios de seguimiento y el desarrollo de tratamientos dirigidos a la EP.
«Al incorporar conjuntos de datos de ascendencia diversa, pudimos acotar las variantes funcionales de algunos de los genes en los loci que identificamos», señala Nacho. «Los datos de ascendencia diversa permiten identificar variantes que podrían ser específicas de una población, por ejemplo, y también permiten identificar variantes que son significativas en determinadas poblaciones y que, por tanto, es más probable que sean factores de riesgo causales importantes».
Algunos de los loci identificados tuvieron efectos diferentes en distintas poblaciones, lo que pone de relieve la importancia de los estudios genéticos que tienen en cuenta factores de riesgo específicos según la ascendencia. Dos loci detectados únicamente en poblaciones latinoamericanas y africanas estaban cerca de los genes JAK1 y HS1BP3. Ambos genes participan en la señalización inflamatoria y pueden apoyar el papel de la inflamación en la EP.
Repercusiones y nuevos horizontes de cara al futuro
Los resultados del estudio subrayan la importancia de incluir diversas ascendencias en la investigación genética. Esta inclusión mejora la precisión de la predicción del riesgo genético y el desarrollo de tratamientos personalizados. El GP2 se ha asociado con instituciones que trabajan con minorías poblacionales para generar datos sobre comunidades desatendidas de todo el mundo.
«Contamos con personas de todo el mundo trabajando en colaboración y aprendiendo unos de otros», expresó Nacho. «Para mí, esta colaboración es lo más emocionante de este estudio y de las posibilidades de cara al futuro».
Se necesitarán más estudios de laboratorio para confirmar cómo los loci específicos identificados en este estudio afectan las vías de la EP y las manifestaciones de la enfermedad. No obstante, este trabajo sienta unas bases sólidas para futuros estudios.
Este estudio ha contado con el apoyo de: Intramural Research Program de los Institutos Nacionales de la Salud (NIH), Instituto Nacional Sobre el Envejecimiento (NIA), NIH, Department of Health and Human Services; National Institute of Neurological Disorders and Stroke; Parkinson’s Foundation; Michael J Fox Foundation; Aligning Science Across Parkinson’s Global Parkinson’s Genetic Project (ASAP-GP2); American Parkinson’s Disease Association; National Medical Research Council de Singapur y Academic Research Fund del Ministerio de Educación de Singapur.
Esta investigación se llevó a cabo usando el UK Biobank Resource, número de solicitud 33601. Queremos expresar nuestro agradecimiento a los participantes e investigadores del estudio FinnGen. Damos las gracias también a los participantes en la investigación y a los empleados de 23andMe. Los datos utilizados en la preparación de este artículo pertenecen al Global Parkinson’s Genetics Program (GP2). El GP2 es un programa financiado por la iniciativa Aligning Science Across Parkinson’s (ASAP) e implementado por The Michael J. Fox Foundation for Parkinson’s Research (https://gp2.org).