
GWASを理解する…そして一体なぜオッズ比がこんなに小さいのでしょうか?
以下の説明は、GWAS研究を非常に単純に表現しており、好意的な皮肉も含んでいます。
ゲノムワイド関連解析(GWAS)によって、病気について何が分かりますか?なぜゲノムの100万を超える独立機能をテストし、深刻な多重検定の乱用に耐えるのですか?答えは非常に単純です。2つの非常に単純な理由があります。まず、遺伝子のすべての候補関連領域(別名遺伝子座)は、疾患に関する知識の範囲を広げます。第2に、観察されるこれらの累積的に小さな影響のすべては、それぞれが遺伝的リスクのわずかな増加をもたらし、リスクをよりよく予測し、診断基準を洗練するのに役立ちます。GWASには仮説がないと言われていますが、実際は「ゲノム内の変異種または変異種群が何らかの悪影響をもたらす可能性があるため、詳細を調べる必要がある」というのが仮説です。
最も基本的なレベルでは、GWASは一連の方程式であり、それぞれが1度に1つのゲノム変異種を調べます。パーキンソン病のコンテクストでこれを分解してみましょう。
パーキンソン病(はいの場合は1、いいえの場合は0) ~ β0 + β1variant + β2sex + β3age + β4PC1 + β5PC2
統計に精通している方には、標準のGWAS式は単純な重回帰式です。この特定の方程式は、その人のゲノム内の特定変異種の存在を使って、パーキンソン病にかかっているかどうかを予測できるかどうかをテストしています。テストされた変異種に加えて、性別、年齢、個人レベルの遺伝的祖先を説明する主成分などの他の要因が含まれます。
それでは、オッズ比とベータについて話しましょう。上記の式では、変数の前の‘βn’項は係数です。これらは、パーキンソン病の予測可能性または「リスク」が、その変数に応じてどの程度増加または減少するかを示しています。GWAS稿では、これらの変異種効果サイズがベータ値またはオッズ比として報告されます。両方を解釈するための例を見てみましょう。これまでの最新かつ最大のPDGWAS(Nalls, 2019)では、PDと有意に関連していることが判明した新しい変異種の1つは、第2染色体上の変異種rs76116224です。この変異種に関連付けられているベータ値は0.110です。オッズ比は1.12です(信頼区間: 1.08–1.16)。1.08–1.16).ベータ値は、結果のユニットあたりの増加または減少です。したがって、この研究では:
- rs76116224変異種の1つのコピーでは、パーキンソン病のリスクが0.110 * 1 = 0.110増加すると推定されています。
- rs76116224変異種の2つのコピーでは、パーキンソン病のリスクが0.110 * 2 = 0.220増加すると推定されています。
オッズ比(OR)はベータ係数の指数ですが、解釈が少し異なります。1より大きいORは結果のより高いオッズに関連付けられ、1のORは関連付けがないことを意味し、1未満のORは結果のより低いオッズに関連付けられます。したがって、ORが1.12の場合、変異種コピーが1単位増加すると、パーキンソン病になるオッズが12%増加すると予想されます。
これで方程式が完成したので、データ内のすべての変異種について方程式を計算する必要があります。したがって、データの処理方法にもよりますが、約25,375,550回です。幸い、これらすべての方程式を実行する優れたソフトウェアがあります。
通常、各GWASは、祖先ごと、および個々のコホートごとに、独自のデータに基づいて別々のセンターで行われます。ただし、考慮する人数が多いほど、希少な効果の変異種を検出する可能性が高くなります。この数を増やすために、各GWASはメタ分析で結合されます。共同サイトは、病気やその他の結果がどのように定義されるか、どのコードと計算インフラを使うか、場合によっては、結果発表論文の著者の順序についても事前に合意します。各研究サイトは通常、研究レベルのジェノタイピング配列データの基本的な品質管理(QC)パラメーターと、コンセンサスの代入方法(同じ参照パネルを使用して分析されていない遺伝的変異を推測する)に同意します。選択した品質管理パラメーターを、GWAS実行前に行う必要があります。
QCが完了し、個々のGWASが実行されたら、基本統計からメタ分析手法を使用して、GWAS要約統計を変異種ごとに1つの集計結果セットに結合します。ここから、いくつかの追加QCの後、p値が5e-08未満の変異種を探し始めます(このカットオフは通常、GWASが数100万の複数テストを修正するために選択され、さらに厳格です)。この有意性のしきい値を超える変異種は、テストされた結果と有意に関連していると見なされますが、これらの結果を追加のデータセットに複製することを試みることが重要です。
GWASによって発見された変異種は、病気に関する知識を調査し、向上させるためのターゲットを提供します。すべての新しいターゲットは、特定の結果を生み出す人のリスクを正確に予測する能力の構築に役立ちます。これらの知見は、治療における精密医療アプローチに移行する際に必要になります。GP2では、パーキンソン病に関する知識の拡大に取り組んでいます。GWASプロセスの多くは自動化されており、使用するコードは透明性をもって公開され、拡大し続けています。私たちは多様性に重点を置いているため、データのグローバルなサイロ全体にわたる統合分析を扱い、可能な場合は調和された統一コホートとして分析し、適切な場合はメタ分析を行います。
各国の多くの研究者や研究機関で使用されることを目指して、一貫した明確なワークフローとコードを構築するために多大な努力を払っています。私たちの目標は、GP2を地球上で最も透明性の高いオープンサイエンス分析プロジェクトにすることです。
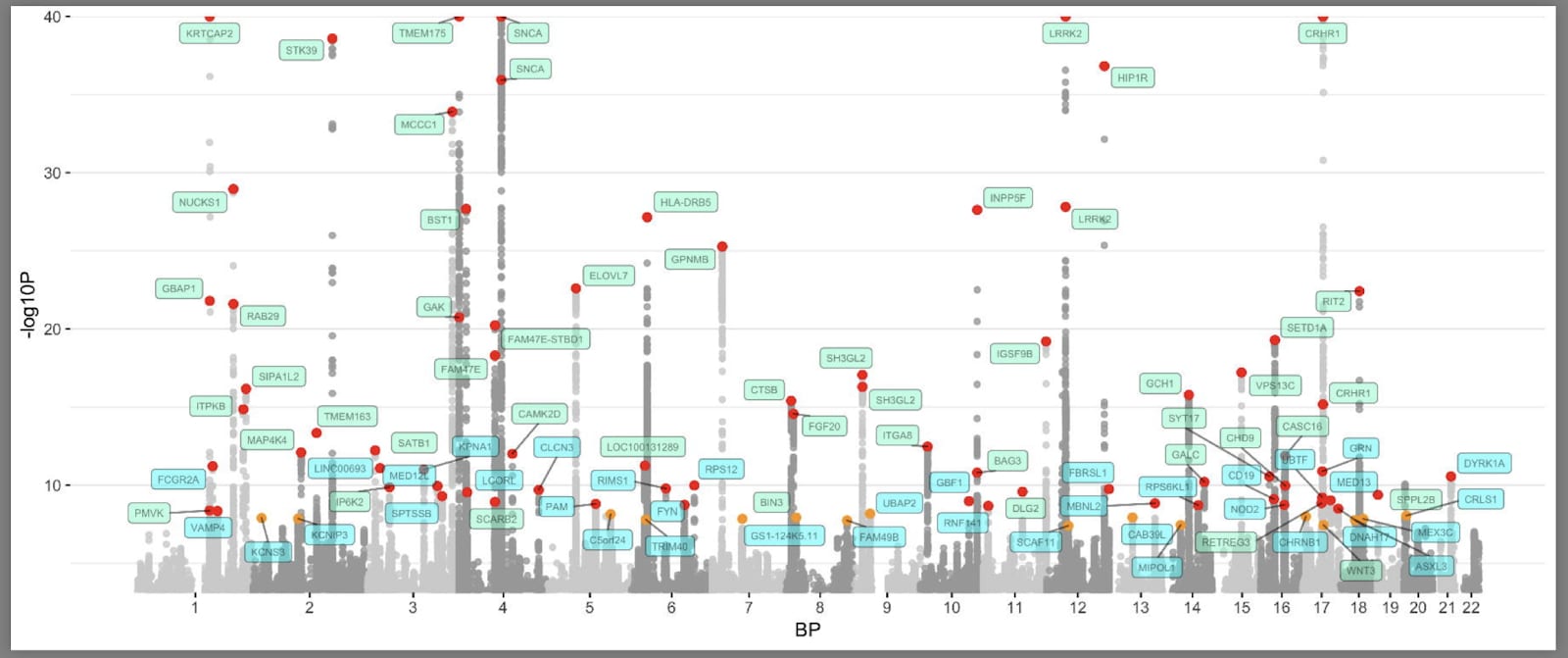
GWASの現在の「マンハッタンプロット」(マンハッタンのタワーにちなんで命名)は上記のとおりです(Nalls et al., 2019)。こちらでコードを見ることができます。
コードとパイプラインはGP2 GitHubにあります。ご自分のGWASを実行するための洞察やヒントを提供するフォローアップブログも探してみてください。


