
前回のブログ記事では、GWASについて、統計的手法やワークフロー、最新のパーキンソン病GWASの例などをご紹介しました。今回は、独自のGWASを実施するための追加的な洞察とヒントを提供したいと思います。
品質管理およびデータの補完
- 高品質な遺伝子型(非パリンドローム性、すなわちA/Tがなく、可能であればG/Cがあり、クラスターの品質を示すイルミナジェントレインスコアが高い)で、欠損率が低いです(サンプルおよびSNPごとに1%未満が望ましい)。
- 一般的には、共通のマイナー・アレル頻度が1%以上であることに着目しますが、それ以下の場合は、遺伝子型クラスタが非常に良く見えることを期待します(検査する必要あり)。
- ヘテロ接合性の高いサンプルは、汚染の可能性があるため、確認してください。また、ヘテロ接合率が低い場合も問題となります。
- サンプルが重複していたり不可解な形で関連しているサンプルには注意が必要です。
- アルゴリズムの観点からサンプルの遺伝的系統を確認し、解析します(我々はfastStructureとflashPCAの組み合わせを使用)。多様性を最大化する一方で、人口の下部構造とその分析結果への影響に注意しましょう。
- 対照の状態やハプロタイプによる欠失によるバイアスを含め、偶然では欠失しないSNPを調べます。
- 1つの集団に対して複数のインプテーション・リファレンス・パネルを使用し、その結果得られた遺伝子型の中で最も品質の高い指標を使用できない理由はありません。これは、混合サンプル集団では特に有効です。
研究レベルの分析のヒント
- 回帰-GWASでは一般的に、連続した成果には線形回帰モデルを、離散した成果にはロジスティックを使用します。これらは、共変量を考慮しながらSNPでのリスクを定量化するものです。
- 様々なソフトウェアパッケージを使用して混合線形モデルを実行することができ、これらのモデルは近縁性や微細な集団の下部構造を正確に説明することができます。
- PC – ゲノムワイドジェノタイピングデータに基づいて主成分負荷を計算することで、回帰モデルの共変量として有効なセットを作成し、集団の下部構造を効果的に考慮することができます。
それではメタ分析しましょう。
- 固定 vs. ランダム – これらは、データサイロや出版物をまたいで、サマリー統計レベルでデータを組み合わせるメタ分析手法です。固定効果メタ分析はディスカバリー分析に役立つことが多く、一般的にはより強力ですが、ランダム効果モデルの方がより保守的で、データサイロ間の異質性を考慮できるため、ランダム効果モデルを好む人もいます。
- 異質性の推定値 – Cochrane’s Q や I2 で表されることが多いです。外れ値効果の偏りや結果の一般化を知る上で、非常に重要です。
精査の結果
- スタディレベルラムダ – 0.95から1.05の間で維持し、それ以外は問題です。
- ラムダ全体およびラムダ1000 – 研究レベルまたはメタ分析レベルでのラムダの計算は、症例がある場合はコントロールの不均衡の都合上厄介です。コントロールの数が非常に多いと、ラムダ統計が人為的に増大することがあります。この場合、1000症例と1000対照にスケールされるので、lambda1000を使用します。繰り返しになりますが、このコードはGitHubのレポにあります。
- Linkage disequilibrium (LD) スコアのインターセプト – ラムダの代替品/補完品で、LD構造や症例に対してより頑強です。つまりコントロールの不均衡です。
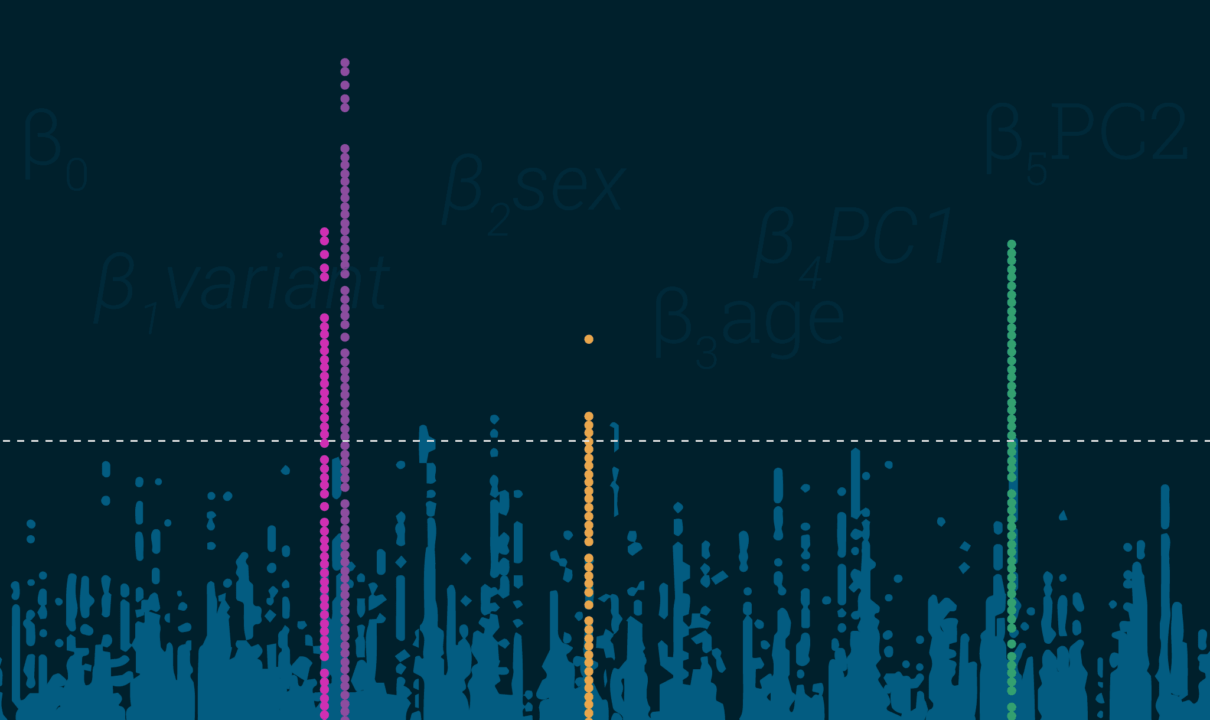
- 均一性を示すプロットのLDピーク – これは非常に簡単なことで、プロットされた結果は、吹雪ではなく、マンハッタン(またはその他の都市、ごめんなさい、ワシントンD.C)のタワーのように見えるはずです。
- 複製 – あるに越したことはないですが、利用可能なデータセットがない場合もあります。追加データが入手できない場合は、leave-one-outメタ分析とクロスバリデーションを組み合わせてみてください。
GWAS後の分析
- あるいは「GWASメタ分析が完了したら何をすべきか」。なお、GP2では、これらのテーマに関するブログ記事や講座を今後も提供していく予定です。
- 条件付き解析 – 地域で最も重要なSNPを共変数として使用し、解析を再実行するか、GCTAのようなツールを使用する。1つの位置に対し複数の独立した信号がある場合もあります。
- ファインマッピング – これは、ベイズコローカリゼーション(遺伝子発現または同様のメトリックのためのゲノム参照データを活用する)を含むcolocのようなパッケージで近似ベイズ因子分析によって行うことができます。
- 予測ウェイトとTWAS – 遺伝子発現、メチル化、クロマチン研究などから得られた外部ウェイトを活用して、GWASデータに隠れている推定上のメカニズムに関連する遺伝子を特定するための様々なパッケージが存在します。
- ML – 目的に応じた予測、私たちの気に入っているパッケージはGenoMLです。というのもそのほとんどが自動化されており、遺伝学における機械学習(ML)を簡単に行うことができるからです。
詳細については、GP2のGitHubにあるコードとパイプラインを参照してください。
このブログはHampton Leonard、Mike Nalls、Yeajin Song、Dan Vitaleにより共同執筆されました。彼らの詳細については、GP2の難病 – データ分析ワーキンググループのページをご覧ください。


