
在上一篇 博文里,我们介绍了全基因组关联研究(GWAS),包括其统计公式、工作流程以及最新的帕金森病GWAS实例。今天介绍更多的见解和技巧,供您在运行您自己的GWAS时参考。
质量控制和估算过程
- 低缺失(优先选择每个样本和每个SNP<1%的)的高质量基因型(非回文,即如果可能的话,没有 A/T,G/C,具有表明集群质量的高Illumina Gentrain分数)。
- 通常关注常见的、次要的>1%的等位基因频率,低于此值则希望基因型簇看起来很好(记得检查)。
- 检查样品是否存在高杂合率,因为这可能表明存在潜在污染。杂合率过低也可能是个问题。
- 注意重复的样本和关联机制不明的样本。
- 通过遗传祖先算法确定和解析样本(我们使用fastStructure 和flashPCA的组合)。最大限度地提高多样性,但要警惕群体子结构及其对分析结果的影响。
- 查看并非偶然缺失的SNP,包括因缺失导致的偏见:对照状态或单倍型。
- 您完全可以对单个群体使用多个插补参考面板,并使用由此得到的具有最佳质量指标的基因型。这在混合样本群体中特别有用。
研究级别分析贴士
- 回归-GWAS通常对连续结果使用线性回归模型,对离散结果使用逻辑回归模型。这就量化了SNP的风险,同时考虑了协变量。
- 多种软件包可用于运行混合线性模型,这些模型可以准确地说明相关性和精细的人口子结构。
- PCs-基于全基因组基因分型数据,计算主成分负荷,为您的回归模型提供一组有用的协变量,让您有效地考虑人口子结构。
现在让我们来进行元分析。
- 固定与随机——这些是元分析方法,用于跨数据孤岛和/或出版物合并数据在汇总统计级别合并数据。固定效应元分析通常对发现分析很有用,而且通常更强大,尽管我们中的一些人更喜欢随机效应模型,因为后者更保守,并且可以解释数据孤岛之间的异质性。
- 异质性估计——通常表示为Cochrane的Q或I2。这很重要,能让我们一窥离群效应偏差和结果的普遍性。
审查结果
- 研究级别拉姆达-将其保持在0.95和1.05之间,超出这一区间都会有问题。
- 总体拉姆达和拉姆达1000——当对照研究不平衡时,在研究级别或元分析级别计算拉姆达可能会很棘手。大量过量的控制会人为地拉高拉姆达统计量。在这种情况下,请使用拉姆达1000,因为它扩展到了 1000个案例和1000个。再次提醒,此对照组代码在我们的GitHub存储库中可以找到。
- 连锁不平衡(LD)得分截距-拉姆达的替代/补充,对 LD 结构和对照研究不平衡更加稳健
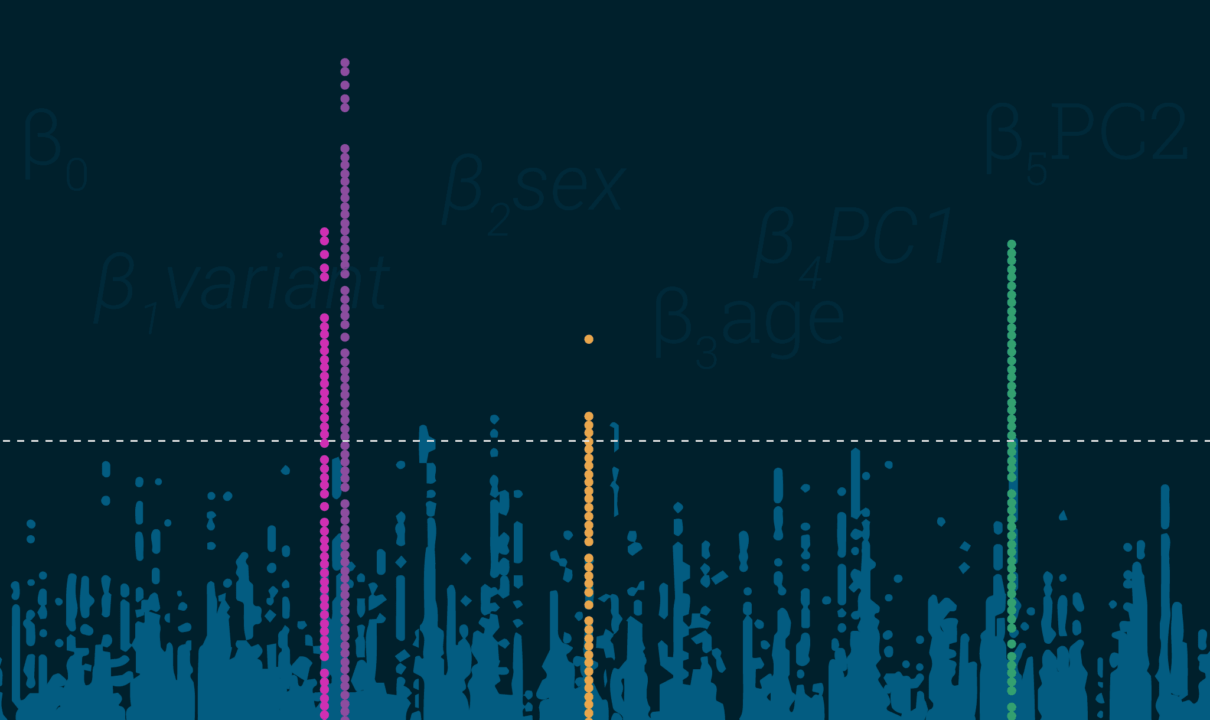
- 图中显示均匀性的LD峰值——这非常简单,绘制时的结果应该看起来像曼哈顿的塔(或是您选择的城市,对不起了华盛顿),而不是像暴风雪。
- 复制——总是好的,但有时你没有可用的数据集。当无法获得额外数据时,尝试结合使用留一法荟萃分析和交叉验证。
一些后GWAS分析
- 或者说“完成GWAS元分析后要做什么?”请注意,GP2后续会就这些话题提供更多的博文和课程。
- 条件分析——使用区域中最重要的SNP作为协变量,并再次进行分析或使用GCTA等工具。每个基因座可能有多个独立的信号。
- 精细映射——这可以通过}包括贝叶斯共定位(利用基因组参考数据进行基因表达或类似指标)的诸如coloc 的软件包中的近似贝叶斯因子分析来完成。
- 预测权重和TWAS——有很多不同的软件包可利用基因表达、甲基化或染色质研究等外部权重来识别隐藏在GWAS数据中的假定机械连接基因。
- ML——专门构建的预测,我们最喜欢的软件包是 GenoML ,因为它基本实现了自动化,让基因组学中的机器学习(ML)变得容易。
如欲获取更多信息,可在 GP2 GitHub上找到我们的代码和流水线。
本博文由Hampton Leonard、Mike Nalls、Yeajin Song和Dan Vitale共同撰写。请访问GP2的复杂疾病-数据分析工作组页面了解作者的背景。



