
In unserem letzten Blogbeitrag haben wir eine Einführung in die GWAS gegeben und dabei statistische Formeln, Arbeitsabläufe und Beispiele aus der jüngsten GWAS für die Parkinson-Krankheit vorgestellt. Nun möchten wir Ihnen zusätzliche Einblicke und Tipps für die Durchführung Ihrer eigenen GWAS geben.
Qualitätskontrolle und Imputationsverfahren
- Qualitativ hochwertige Genotypen (nicht-palindromisch, d. h. möglichst kein A/T, G/C, mit hohem Illumina Gentrain Score zur Clusterqualität) mit geringer Missingness (vorzugsweise < 1 % pro Probe und pro SNP).
- Konzentrieren Sie sich grundsätzlich auf verbreitete Frequenzen des selteneren Allels (Minor Allele Frequencies, MAF) > 1 %, bei geringerem Wert ist zu hoffen, dass das Genotyp-Cluster gut aussieht (sollte überprüft werden).
- Überprüfen Sie die Proben auf hohe Heterozygotieraten, da dies auf eine mögliche Kontamination hinweisen könnte. Niedrige Heterozygotieraten können ebenfalls problematisch sein.
- Vorsicht bei duplizierten Proben und kryptisch verbundenen Proben.
- Überprüfen und parsen Sie die Proben algorithmisch auf genetische Abstammung (wir arbeiten mit einer Kombination aus fastStructure und flashPCA). Die Vielfalt sollte möglichst groß sein, dabei jedoch die Substruktur der Bevölkerung und deren Auswirkung auf die Analyseergebnisse berücksichtigt werden.
- Achten Sie auf SNPs, die nicht zufällig fehlen, einschließlich Verzerrungen durch Fehlen bei Fall-Kontroll-Status oder Haplotyp.
- Es gibt keinen Grund, warum man nicht verschiedene Imputationsreferenzpanels für eine einzige Population verwenden und den Genotyp mit der besten Qualitätsmetrik auswählen sollte. Dies ist besonders nützlich bei gemischten Stichprobenpopulationen.
Tipps zu Studienniveauanalysen
- Regression – Bei der GWAS werden zumeist lineare Regressionsmodelle für kontinuierliche Ergebnisse und logistische Modelle für diskontinuierliche Ergebnisse verwendet. Diese quantifizieren das Risiko bei einem SNP unter Berücksichtigung von Kovariaten.
- Mit verschiedenen Softwarepaketen können gemischte lineare Modelle ausgeführt werden, bei denen Verwandtschaft und feinskalige Populationssubstrukturen genau berücksichtigt werden können.
- Hauptkomponenten – Durch die Berechnung von Hauptkomponentenladungen auf der Grundlage genomweiter Genotypisierungsdaten erhalten Sie einen nützlichen Satz von Kovariaten für Ihre Regressionsmodelle, dank derer Sie die Substruktur der Population berücksichtigen können.
Lassen Sie uns nun eine Metaanalyse durchführen
- Fixed vs. Random – Dies sind Meta-Analyse-Methoden zur Kombination von Daten auf der Ebene einer zusammenfassenden Statistik, über Datensilos und/oder Publikationen hinweg. Fixed-Effects-Metaanalysen sind oft nützlich für entdeckende Analysen und im Allgemeinen aussagekräftiger. Manche arbeiten jedoch lieber mit Random-Effects-Modellen, da sie konservativer sind und die Heterogenität verschiedener Datensilos berücksichtigen.
- Schätzung der Heterogenität – Häufig ausgedrückt in Cochrane’s Q oder I2. Sehr wichtig, denn dadurch erhält man einen Eindruck von der Verzerrung durch Ausreißereffekte und von der Verallgemeinerbarkeit der Ergebnisse.
Ergebnisse der Prüfung
- Lambda-Werte in der Studie – sollten zwischen 0,95 und 1,05 liegen, ansonsten hat man ein Problem.
- Gesamt-Lambda und Lambda 1000 – Die Berechnung von Lambda-Werten auf Studien- oder Metaanalyse-Ebene kann schwierig sein, wenn ein Fall-Kontroll-Ungleichgewicht besteht. Durch exzessive Kontrollen kann die Lambda-Statistik künstlich aufgebläht werden. Verwenden Sie in diesem Fall Lambda 1000, denn der Wert ist auf 1000 Fälle und 1000 Kontrollen bezogen. Wie gesagt, der Code hierfür findet sich in unserem GitHub Repository.
- Linkage Disequilibrium (LD) Score Intercept – Alternative/Ergänzung zum Lambda-Wert und robuster gegenüber der LD-Struktur und dem Fall-Kontroll-Ungleichgewicht.
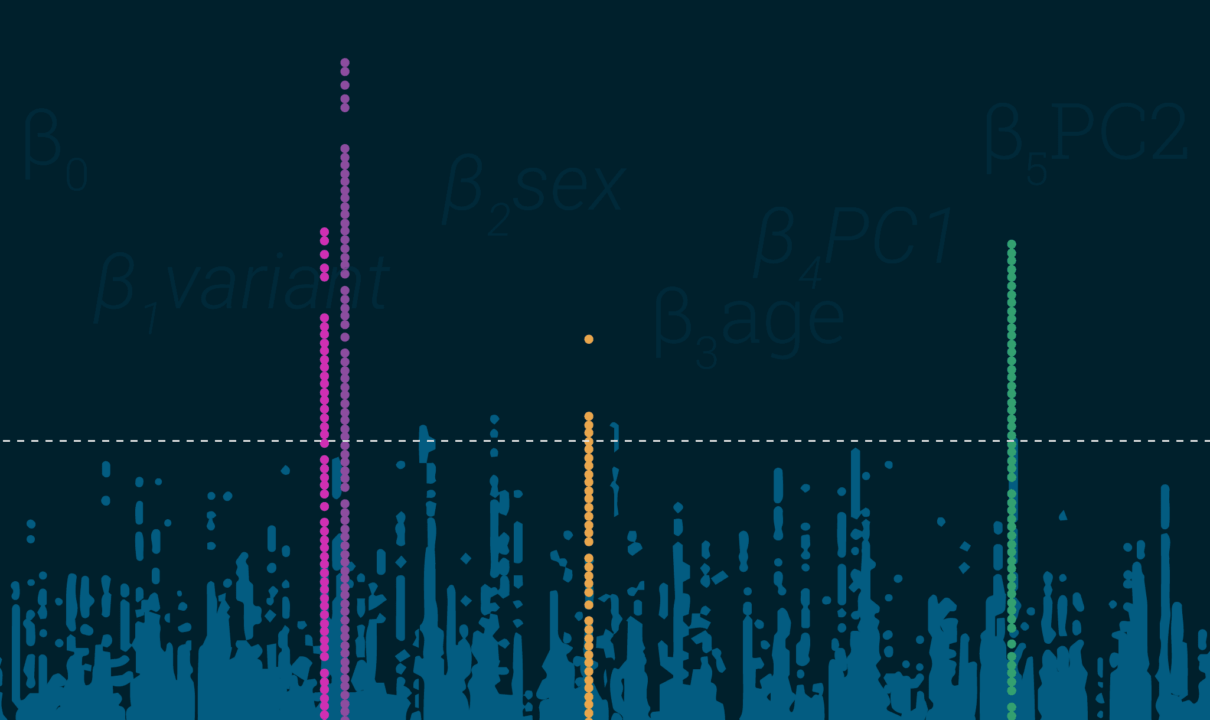
- LD-Spitzen in gleichförmigen Punktdiagrammen – das ist relativ eindeutig. Ihre Ergebnisse sollten als Punktdiagramm wie die Skyline von Manhattan (oder die Skyline der Stadt Ihrer Wahl – Sorry, D.C.!) aussehen, nicht wie ein Schneesturm.
- Replikation – immer gut, aber manchmal hat man keine Datensätze zur Verfügung. Wenn keine zusätzlichen Daten verfügbar sind, versuchen Sie es mit einer Kombination aus Leave-One-Out-Metaanalyse und Kreuzvalidierung.
Einige Analysen nach der GWAS
- Oder: „Was kommt nach der GWAS-Metaanalyse?“ Zur Information: GP2 wird weitere Blogbeiträge und Kurse zu diesen Themen anbieten.
- Bedingte Analysen – Verwenden Sie den signifikantesten SNP in einer Region als Kovariate und führen Sie die Analysen erneut durch oder verwenden Sie das Tool GCTA. Eventuell gibt es mehrere unabhängige Signale pro Locus.
- Feinkartierung – Dies kann durch eine approximative Bayes-Faktor-Analyse in Paketen wie coloc einschließlich Bayesscher Kollokation erfolgen (unter Nutzung von genomischen Referenzdaten für Genexpression oder ähnlichen Messgrößen).
- Prognosegewichtung und TWAS – Es gibt eine Reihe von Paketen zur Nutzung externer Gewichtungen aus Genexpressions-, Methylierungs- oder Chromatinstudien usw., um mutmaßlich mechanistisch verbundene Gene zu identifizieren, die sich in Ihren GWAS-Daten verstecken.
- ML – Zweckgebundene Prognosen; unser Lieblingspaket ist GenoML, denn es ist weitestgehend automatisiert und macht maschinelles Lernen (ML) in der Genomik einfach.
Zur weiteren Information finden sich unser Code und die Pipelines im GP2 GitHub.
Dieser Blogbeitrag wurde gemeinschaftlich von Hampton Leonard, Mike Nalls, Yeajin Song und Dan Vitale verfasst. Wenn Sie mehr über die Autoren erfahren möchten, besuchen Sie gerne die Webseite der GP2 Complex Disease – Data Analysis Working Group.

